
Abstract
Gene therapy has emerged as a promising therapeutic avenue, offering targeted treatments for various diseases. Purification of viral vectors presents a pivotal challenge, demanding the removal of impurities while preserving integrity and potency. During manufacturing, producer cells in transfection systems can be transiently transfected or retro-infected by the viral vectors they have just produced—a process referred to as “retro-transduction”—leading them to express the transgenes of interest. This can be a significant source of contamination in the viral solution pool, particularly when the transgenes encode extracellular, secreted proteins, resulting in cytotoxicity and reduced viral potency. Herein, we aimed to evaluate the efficiency of different viral purification systems commonly used in academic and industry settings in removing the transgene background from viral solutions. The efficiency of each system was assessed based on the levels of the secreted transgene Gaussia Luciferase (GLuc), which can be quickly detected in a solution and served as a readout for transgene background contamination in the viral pool during downstream processing. Through a systematic evaluation of purification methods, we identified the most effective approaches for producing pure viral batches with minimal transgene background, all while preserving viral potency and functionality. Our study revealed superior performance of batches that underwent purification via tangential flow filtration, which yielded over 90% reduction in GLuc background and the highest transduction efficiency rates. This work provides significant insights for advancing gene therapy applications that rely on the production of viral vectors encoding secreted transgenes.
INTRODUCTION
In recent years, gene therapy has emerged as one of the most promising therapeutic approaches with the potential to treat a broad spectrum of diseases with high specificity and long-lasting effects. 1,2 Viral vectors such as lentiviruses (LVs) are invaluable tools in gene therapy, serving as competent genetic material delivery platforms, providing efficient, stable, and long-term transgene expression in several target tissues. 3 –6 Despite the progress achieved in the area, viral vector manufacturing capabilities have fallen behind, especially at a research scale. 7,8 Academic facilities are usually restricted to the initial stages of gene therapy clinical development due to scalability challenges, inefficient downstream purification processes, supply chain limitations, and regulatory requirements.
Viral vector manufacturing is a field with many trade secrets and high variability. Unlike conventional small-molecule drugs, biopharmaceutical manufacturing can face challenges at essentially every step. For instance, purification of viral batches is a critical step to remove impurities and contaminants while preserving vector potency and integrity. 9,10 That step is especially crucial for producing viral vectors that encode secreted transgenes. During viral manufacturing, producer cells can be transiently transfected or retro-infected by the viral particles they have just produced and released into the supernatant, leading to the unintended stable transduction of these cells—a process known as “retro-transduction.” 11 These mechanisms lead to the expression of genes of interest by the producer cells and, in the case of extracellular proteins, result in the improper secretion of these proteins into the viral pool supernatant.
Transduction of producer cells can lead to the loss of up to 90% of viable viral particles, significantly reducing viral titers and overall production efficiency. 12,13 Undesired transgene contaminants may remain in the viral solution even after downstream processing (DSP) and purification, directly impacting viral integrity and potency. Furthermore, if the resulting viral solution is not adequately purified of transgene background and contaminants, it can negatively impact transduction efficiency, as therapeutic transgenes may be cytotoxic or even induce apoptosis of target cells. 14 This transgene background can also increase the complexity of quality control of viral batches, raising safety concerns, such as variable efficiency and increased risk of immunogenicity, adding time to the manufacturing process. That becomes particularly important in larger production scales for in vivo applications, especially in academic settings where purification capabilities are more limited.
In this study, we aimed to evaluate viral purification methods commonly used in academia and industry, exploring various approaches applicable to different viral production scales. Importantly, the elimination of secreted transgene contaminants from the viral batch was assessed using the secreted biomarker Gaussia Luciferase (GLuc) that can be easily detected in solution. 15,16 Finally, the transduction efficiency of each viral batch was compared to determine whether minimizing transgene background resulted in differences in viral potency and effectiveness. Overall, we evaluated purification systems that facilitated the production of pure viral batches with reduced secreted transgene contaminants, making them particularly suitable for gene therapy applications involving extracellular therapeutic transgenes.
MATERIALS AND METHODS
Lentiviral transient transfection: General reagents and procedures
Human embryonic kidney cell line HEK293T (ATCC CRL-3216) was expanded for 2 days in T175 flasks in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) medium with 10% fetal bovine serum (FBS) (Gibco) and 1% antibiotic/antifungal (ThermoFisher). The HEK cell line starting stock always ranged from passages 6–10. Cells were trypsinized using 1× trypsin in Phosphate-Buffered Saline (PBS) and centrifuged at 300 g for 5 min to pellet cells and resuspended in DMEM/F12 complete. Plasmids used for LV transfections were LV-EF1α-RFP, LV-EF1α-GLuc-IRES-RFP, or LV-sGP130-IRES-GLuc, pspax2 (packaging), and pmd2g (envelope) (Addgene), which all contain ampicillin resistance genes. Plasmid maxi-prep stocks were prepared by transforming plasmids in One Shot Stbl3 Chemically Competent E. coli cells (ThermoFisher). Transformed cells were plated on LB agar plates with ampicillin and cultured overnight at 32°C. Colonies were selected to smear on new ampicillin plates and cultured overnight at 32°C. Bacteria was collected from the plates using a pipette tip and transferred to 500 mL liquid Luria-Bertani agar (LB) broth cultures containing 100 μg/mL ampicillin and cultured in a shaking incubator at 225 rpm at 32°C overnight. Plasmid DNA was isolated and purified using the PureLink HiPure Plasmid Maxiprep kit (ThermoFisher) by column purification. DNA was eluted in 500 μL Tris-EDTA Buffer (TE) buffer (10 mM Tris-HCl, 1 mM Ethylenediaminetetraacetic Acid (EDTA), pH 8.0) (ThermoFisher) and quantified. Quantification of DNA concentration was performed using μDrop plates and read using Varioskan LUX (ThermoFisher) plate reader. Preps were ensured to be within a range of 1–2 µg plasmid/mL, minimum. Polyethylenimine (PEIpro, Polyplus-transfection) was used for polyethylenimine in transfections. A reduced serum medium was used to optimize transfection conditions. OptiMEM (ThermoFisher) was prepared without serum or antibiotics and with a pH of 7.2 using sodium bicarbonate. For collection, a rinse step was performed with a 0.001% Pluronic F-68 solution. Supernatant collection after transfection was filtered using 0.45 μM nylon membrane syringe filters (Corning) attached to a 20 mL syringe. For large-scale transfections greater than 100 mL, a 0.22 μM cellulose acetate membrane vacuum filter unit (Corning) was used to filter supernatant.
10-cm and 15-cm dish transfections
For small-scale transfections, 10- or 15-cm diameter culture dishes were used (Corning). Cells were seeded at 70,000 cells/cm2 one day prior to transfection unless otherwise specified. For 10- and 15-cm dish transfections, PEI:DNA solution was made up in 0.5 and 1 mL solutions, respectively. To do so, PEI was added at a ratio of 2.5 μL PEI per 1 μg of DNA. For 10 cm dish transfections, 52.5 μL PEI was added to OptiMEM for a total volume of 0.25 mL. In a separate tube, 21 μg of plasmid DNA total was added to OptiMEM for a total volume of 0.25 mL. Both tubes were mixed by vortexing, and then PEI solution was added to the DNA solution and mixed by vortexing. Masses of DNA for each plasmid were calculated based on plasmid size (base pair length) and concentration to yield a 1:1:1 molar ratio of the transgene, packaging, and envelope plasmids. Plasmid length was converted to a molar concentration using the New England Bio molarity calculator for DNA. The PEI-DNA solution was incubated at room temperature for 15 min to allow complexes to form. Media was replaced in the 10-cm dish with 10 mL of OptiMEM. After incubation, the PEI-DNA solution was added dropwise to the dish, ensuring that all areas of the dish were covered and swirled gently to mix. The same process was used for 15-cm dish transfections, but the values were scaled based on the increase in cell number and surface area.
The following day, media was replaced with 10 mL fresh OptiMEM, and cultured for an additional 48 h before viral particle harvesting. Supernatant was collected, and the dish was rinsed with 5 mL 0.001% Pluronic solution. Both solutions were filtered through a 0.45-μm syringe filter and stored in single-use aliquots at –80°C.
HYPERFlask® transfections
For large-scale transfections, a HYPERFlask® (Corning), which is a 10-layer flask with 1,720 cm2 surface area, was used. One day prior to transfection, 13.8 million cells were seeded in the flask. To do so, 20 mL of cell suspension was added to 500 mL DMEM/F12 complete medium and mixed by stirring in a liter bottle. The solution was then added to the HYPERFlask® by pouring at a 45° angle, according to the manufacturer protocol. Slow pouring was performed to ensure even spreading across all ten layers of the flask and to mitigate foam production. An additional 100 mL DMEM/F12 was added to fill the flask to the top cap thread. The top and bottom two layers of the flasks were checked under the microscope to ensure even cell mixing. The flask was incubated horizontally overnight to allow for cell attachment.
The following day, DNA and PEI solutions were prepared in 10 mL of OptiMEM, and the PEI solution was added to the DNA solution in a 50 mL conical tube. A total of 1,680 μL of PEI was used with a total of 672 μg of DNA. After incubation, the PEI-DNA solution was added to 500 mL OptiMEM in a liter bottle and mixed gently with a 50 mL serological pipette. The HYPERFlask® supernatant was discarded by pouring. The OptiMEM containing transfection complexes was added to the HYPERFlask® by pouring. Cells were cultured for 5 days and the supernatant was collected by pouring. Cells were rinsed with 200 mL of Pluronic solution and collected by pouring.
Lentiviral purification
Sucrose-gradient purification
Viral supernatant was collected from 10 cm dishes and passed through a 0.45-µm filter into a 50 mL conical tube using a 20 mL syringe. A total of 3 mL of 20% sucrose solution was laid in a conical bottom 30 mL ultracentrifuge tube. Very carefully, filtered medium was laid on top of the sucrose solution, and two separate layers were clearly visible. The tube was topped up with fresh DMEM to avoid collapsing during the ultracentrifugation spin. Samples were ultracentrifuged at 75,000 g for 90 min at 4°C. Supernatant was discarded into a bleach pot, and tubes were kept inverted on paper tissue for 10 min. The LV pellet (not visible to the naked eye) was then gently resuspended in 3 mL of sterile PBS.
Lentiviral concentrator
Viral supernatant was collected from HYPERFlask® batches and briefly centrifuged at 500 g for 10 min to remove cells and debris. The clarified supernatant (3 volumes) was combined with 1 volume of Lenti-X concentrator (Takara Bio, Cat #631231). The mixture was incubated at 4°C overnight. Samples were then centrifuged at 1,500 g for 45 min at 4°C. The supernatant was carefully removed, and the pellet was gently resuspended in 3 mL of sterile PBS.
Tangential flow filtration
For batches larger than 100 mL (from either multiple dishes or HYPERFlask®), a tangential flow filtration (TFF) system was used to concentrate and purify LV from the supernatant, as previously described. 17 The Minimate benchtop TFF system (Pall) was optimized for LV concentration. Using a 100 kDa Molecular Weight Cut-Off (MWCO) membrane capsule (Pall), the reservoir was connected with tubing to the membrane capsule to the TFF capsule at the feed entry point, and tubing directed the filtrate to a waste container and the retentate back into the reservoir. Pressure gauges were placed upstream and downstream of the capsule to be monitored during the run. A peristaltic pump was connected to the tubing between the reservoir and the capsule to pump the feed into the system. Clamps were placed on the retentate and filtrate tube so that pressure could be altered and filtrate flow could be shut off during collection. The peristaltic pump was set to 70 rpm to achieve a pressure of 20 psi in the upstream pressure gauge, which is recommended by the user manual. The screw clamp on the filtrate tube was adjusted to maintain a pressure of 20 psi in the downstream tubing as well.
The supernatant and PBS rinse solution were added to the reservoir, and the pump was started. Every 20 min, the reservoir levels were checked, and pump speed was adjusted to maintain 20 psi pressure in the upstream and downstream tubing. When the volume in the reservoir reached 50 mL, the pump was paused, the filtrate flow clamp was closed off, and the retentate flow clamp was loosened entirely so there was no pressure in the retentate tubing. The flow direction was then reversed, and the pump speed was increased to 200 rpm to remove any cake layer on the membrane. This step ensures the viral particles are in solution of the retentate. The clamps were then replaced, and the flow direction was returned until the retentate solution approached 25 mL. The filtrate tube was then clamped off and then the three-way valve on the retentate tube was opened, and retentate was collected. An additional 5 mL of PBS was added to remove the hold-up volume of the system, and the last 5 mL of retentate volume was collected. To flush the system, 200 mL of PBS was added to the reservoir, and the system was pumped in reverse for 15 min and then collected. The total processing time on average was about 90 min and was performed at room temperature.
The system was cleaned using a 0.5 M NaOH solution in diH2O. Briefly, the solution was pumped at 300 rpm with the retentate and filtrate clamps fully loosened for 20 min. The flow was reserved, and fresh solution was pumped for an additional 20 min. All tubing and connectors were removed and rinsed with soap and water and then with 0.5 M NaOH before drying. The membrane cassette was filled with 0.5 M NaOH, closed off with luer caps at all ports, and stored.
Collection, storage, and titration
With final batch products in PBS + Pluronic-F68 solution, 1 mL aliquots of the virus were created and added to cryovial tubes. Product aliquots were preserved by freezing at −80°C for up to 1 year and only thawed once immediately before use. A single aliquot was set aside for (qPCR) Quantitative Polymerase Chain Reaction viral titer quantification so that the titer was determined after one thaw cycle as well. Vector titer was determined by (RT-qPCR) Reverse Transcription qPCR using an LV titration kit (Applied Biological Materials, LV900) on Quant Studio 3 (Thermo Fisher).
Lentiviral transduction
HEK-293T cells (ATCC) were seeded at 25% confluence in 24-well plates and allowed to adhere overnight in DMEM complete (ATCC) with 10% FBS (GIBCO) and 1% antibiotic/antifungal. Lentiviral vectors were added at different multiplicities of infection (MOI) to each well in a total volume of 1 mL using antibiotic-free medium. A total of 20 ng/mL of the transduction enhancer Polybrene (Sigma-Aldrich) was also added to each well. Cells were incubated for 24 h before the medium was changed to complete medium. Post-transduction was initiated at 72 h, and plates were scanned for RFP fluorescence imaging and quantification using the Celigo® Imaging Cytometer (Nexcelom Bioscience, Lawrence, MA). Finally, 300 μL of supernatant was collected from each well for GLuc assessment.
GLuc assay
GLuc’s activity was measured using a bioluminescent flash assay. The substrate, native coelenterazine (CTZ), was reconstituted at 2.7 mg/mL in 200-proof ethanol. Working solutions were made fresh immediately prior to the assay with a 1:1000 dilution in PBS. Twenty microliters of each sample were added to black-walled, clear-bottom 96-well plates and 100 μL of CTZ solution and immediately read using a bioluminescent plate reader (Varioscan, Thermofisher). All samples were read forward and in reverse to account for any time-dependent signal degradation. No more than 12 samples were run at a time. GLuc activity in each sample was calculated by taking the average of the forward and reverse readings and expressed as relative light units.
Statistical analyses
Experimental data were analyzed using GraphPad Prism 9.5.1 for Mac OS X (GraphPad Software, La Jolla, CA). The results are shown as the mean ± standard deviation (SD), and significance is set at p ≤ 0.05. For multiple comparisons, one-way or two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-test was used unless otherwise noted.
RESULTS
Process scale-up: Parameter characterization
For effective process scale-up, a universal metric for productivity must be employed. The number of viral particles produced per cell, for example, is a standard metric that can be used when scaling up. This metric is not commonly reported in literature, which makes optimization parameters hard to compare, but it is reported more frequently in suspension-based transfection cultures. In adherent cell-based systems such as ours, the range of viral production per cell can vary substantially, 18 depending on the viral type/serotype, size of transgene, culture vessel, etc. We sought to determine whether cellular production could be maintained with scale.
First, the total yield was quantified for several transgenes in both 10-cm and 15-cm dish transfections. For reference, 0.506 million cells were seeded in 10-cm dish transfections, and 13.8 million cells were seeded in 15-cm dish transfections, so the yield was expected to be approximately 2.75-fold higher. A total of four transfections were performed for each transgene and dish size, and viral titration was determined via qPCR (Fig. 1A and Fig. 2A). As mentioned, titer can be highly variable depending on the transgene, so we used a wide range of coding sequences, testing standard transgene cassettes as well as larger ones with more than one protein expressed in tandem. The total yield across all transgenes averaged 4–5-fold higher in 15- than in 10-cm dish transfections, which was beyond expected, maybe due to the specific plasmids used. Notably, the construct with the largest payload, encoding a secreted anti-cytokine protein (sGP130), exhibited lower overall yields, aligning with earlier findings in the literature. 19
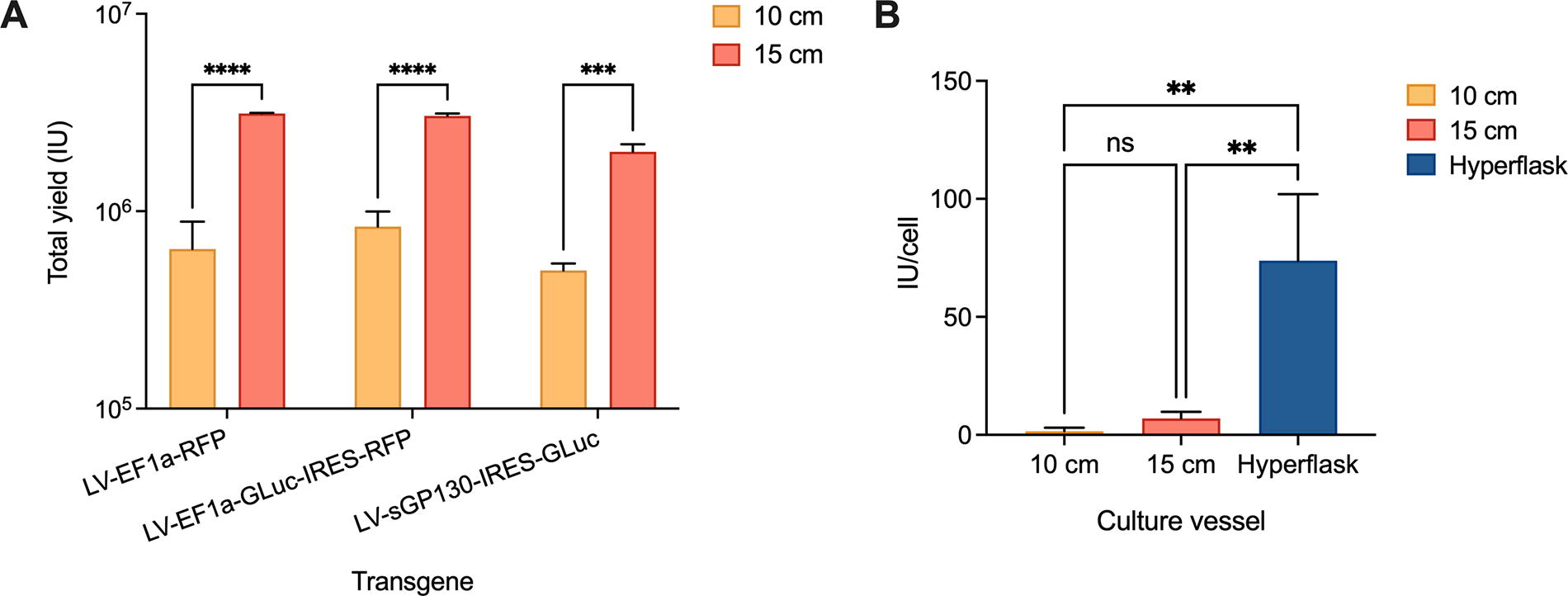
Viral transfection yields vary in different culture vessel scales.

Schematic of the workflow used to evaluate various purification methods.
When normalized for cell seeding number, the average yield per cell did not significantly increase from 10- to 15-cm dish transfections, despite DNA, PEI, and cell number being scaled proportionally (Fig. 1B). When transfections were further scaled up to multilayer cell culture vessels (HYPERFlask®), the average yield exhibited a 10-fold increase compared with 15-cm dishes and an even greater increase compared with 10-cm dishes.
DSP: Minimizing transgene background
Implementing robust purification processes to remove contaminating particles from viral batches is crucial to ensure quality control of these biopharmaceuticals. We next investigated different purification processes to minimize transgene background products in viral batches. For a more precise determination of purification efficiency, LVs expressing a secreted biomarker (GLuc) and a red fluorescent protein (RFP) tag under the strong constitutive promoter EF1α were employed (Fig. 2A). Different production scales were compared (10 cm and HYPERFlask®) and suitable purification methods were employed for each (Fig. 2B). Downstream, the purification efficiency of each method was evaluated based on GLuc background, and viral transduction analyses were carried out for each different batch, as illustrated in Figure 2C.
Ten-cm dish transfections were carried out using a sucrose-gradient purification protocol, a well-documented method in existing literature. 20,21 Batches were analyzed for viral titration before and after purification, as an ideal protocol should result in minimal viral loss. Figure 3A shows an approximate 1.5-fold reduction in the total number of viral particles postsucrose purification, a significant decrease worthy of consideration. Next, GLuc background was determined before and after purification in 10-cm dishes, as depicted in Figure 3B. A significant reduction in the secreted transgene was observed, suggesting good purification efficiency to eliminate impurities and undesirable background.
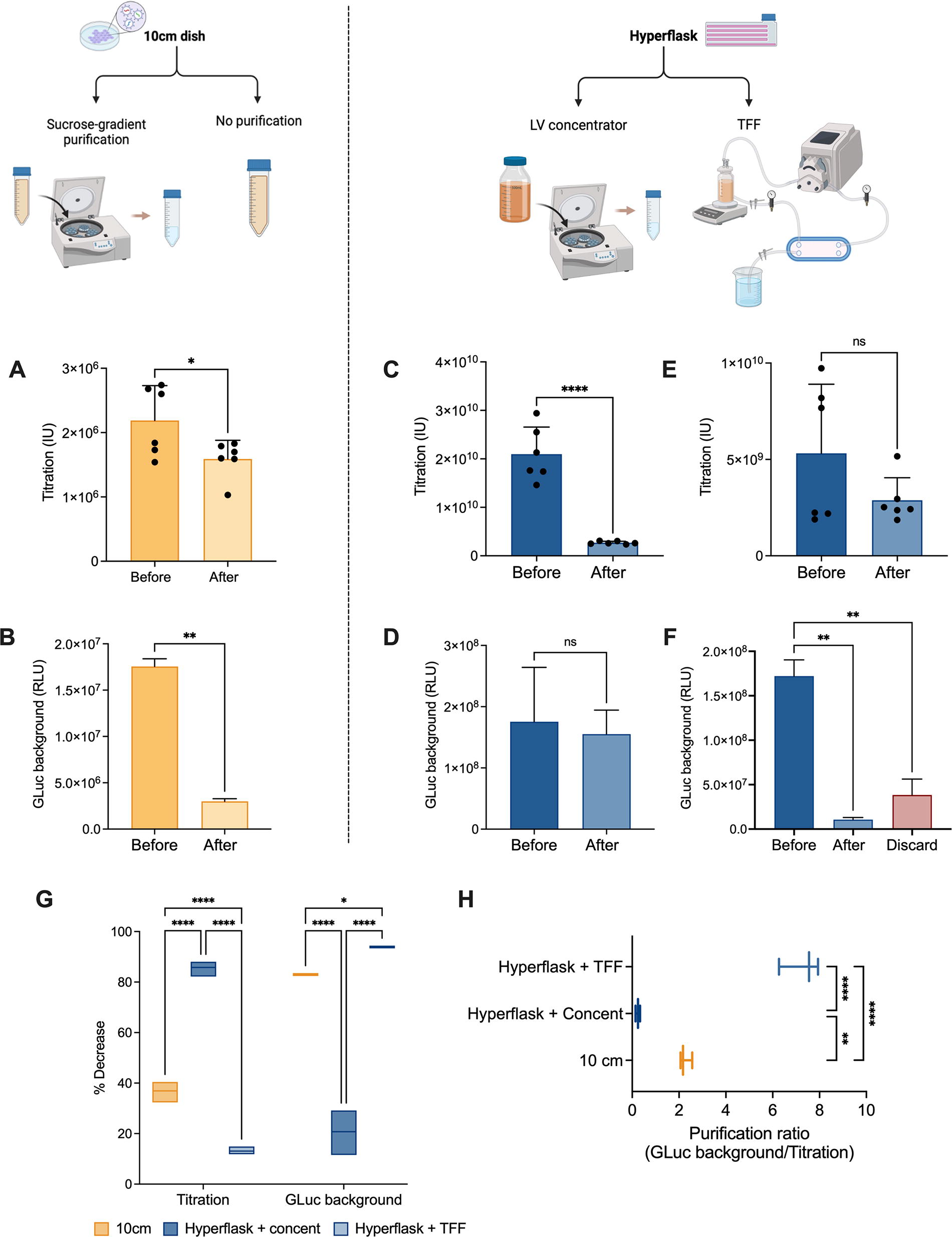
Evaluation of purification efficiency of different methods in minimizing transgene expression background.
HYPERFlask® transfection batches underwent two distinct purification methods: half of them were treated with a commercially available lentiviral concentrator solution, capable of reducing the overall viral suspension volume, enabling the elimination of most residues and debris from the starting supernatant; the other half underwent TFF, a widely used method in bioprocessing and separation technology, that operates on the principle of crossflow filtration, where the feed solution flows tangentially across the surface of a semi-permeable membrane (see Materials and Methods). Both original and concentrated viral solutions were analyzed for final titration and GLuc background. Viral concentration led to a significant loss of viral particles, resulting in an approximately 8-fold decrease in the final count (Fig. 3C). Interestingly, the GLuc background was not significantly reduced, as it would have been expected (Fig. 3D). On the contrary, TFF resulted in the lowest loss of viral vectors while significantly reducing GLuc background by 16-fold (Fig. 3E–F). An optimal purification method should minimize its impact on viral titration while maximizing the reduction in transgene background. Figure 3G depicts the percentage decrease for both parameters, confirming an optimal profile in the HYPERFlask® + TFF group, that is, the lowest impact on titration (less than 15%) and the most significant reduction in GLuc background (over 90%). In addition, when these two parameters were correlated as a purification ratio, the TFF group displayed the highest score, consistent with previous observations (Fig. 3H).
Viral potency assessment post-DSP
Next, the potency and effectiveness of purified and non-purified viral batches were evaluated and compared via viral transduction. First, HEK-293T cells were infected with increasing MOIs of 10-cm viral batches, and transduction efficiency was determined 72 h later. Cells were assessed for RFP fluorescence and GLuc secretion in the supernatant. Importantly, as we transitioned from discussing transfection to transducing target cells, GLuc secretion was now utilized as a readout for transduction efficiency rather than as a “contaminant” parameter. Figure 4A shows increasing RFP fluorescence with increasing MOIs, with a significant difference between purified and non-purified batches (Fig. 4B). In addition, GLuc secretion in the supernatant followed a similar pattern, with a significantly higher expression in batches that underwent sucrose-gradient purification (Fig. 4C). This observation suggests not only the preservation of viral function postpurification but also a potential enhancement thereof.
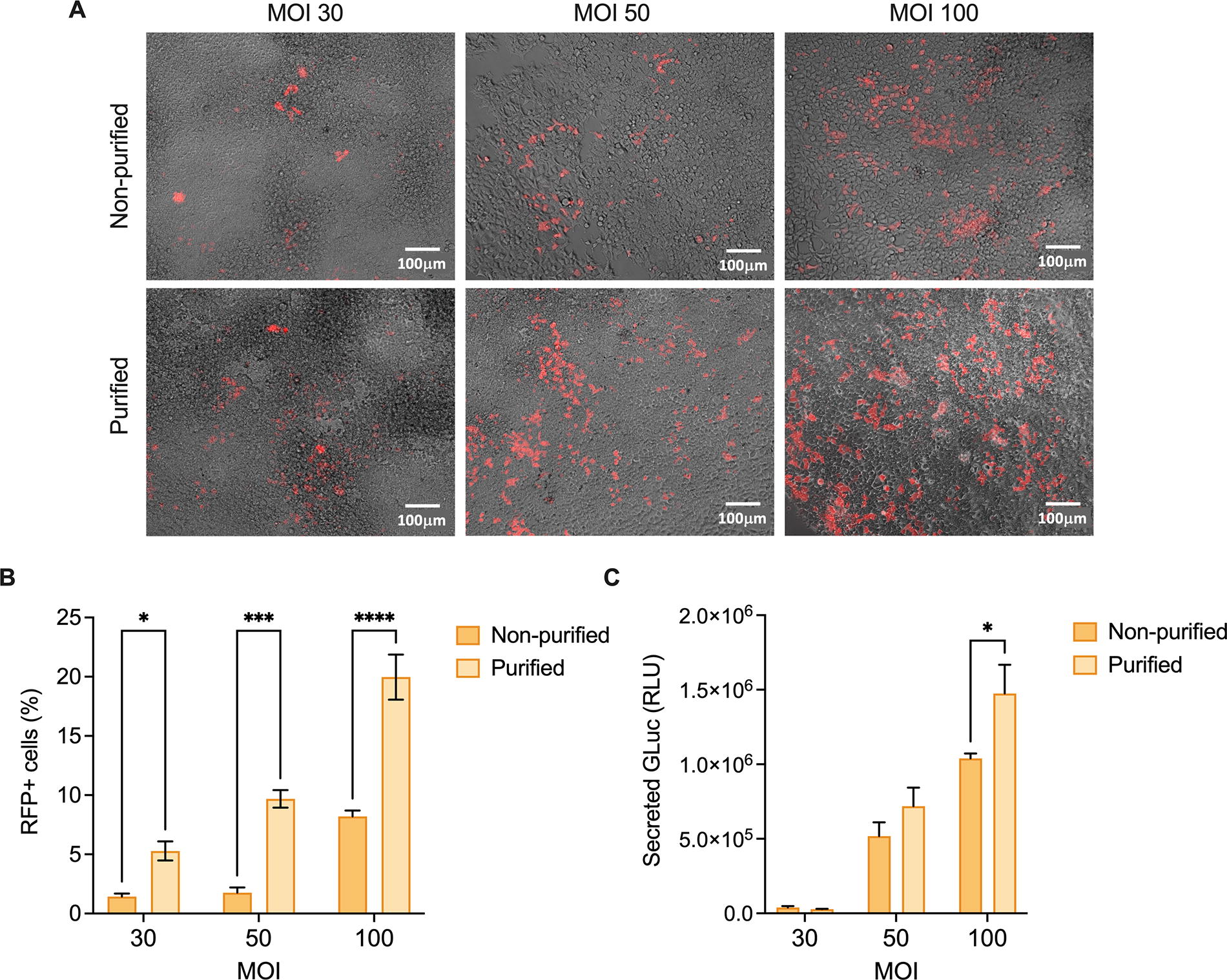
Assessment of viral potency and effectiveness postsucrose-gradient purification.
Finally, viral function was also assessed in purified and non-purified HYPERFlask® batches. Considering the purification ratios previously obtained, only the TFF HYPERFlask® group was tested. Figure 5A shows a marked increase in RFP fluorescence with increasing MOIs, which is substantially more pronounced in the purified group. The quantification of fluorescence in each group confirms that observation (Fig. 5B). Ultimately, GLuc secretion exhibited a parallel trend, with significantly higher expression in groups that underwent TFF purification at MOIs 50 and 100 (Fig. 5C). Noteworthy, the findings here observed mirror the purification efficiency analysis presented in Figure 4, reaffirming TFF as the most effective method for transgene background purification, all while preserving viral potency and integrity.

Assessment of viral potency and effectiveness post-TFF purification.
DISCUSSION
The translation of promising viral vector-based therapies from bench to bedside faces significant challenges, particularly in the production phase. 18,22,23 At the research scale, where innovative therapies are developed and optimized, the manufacturing process is often hindered by numerous bottlenecks that impede efficiency and scalability. 24 This can hinder technological advancements and translation to clinic as in vivo studies require a large number of vectors that are usually beyond the production capabilities of research laboratories. For instance, in vivo experiments in mice typically require lentiviral doses ranging from 1 × 108 to 1 × 109 IU per animal. 25,26 A standard research lab usually produces between 1 × 106 and 1 × 107 IU/mL in one standard 15 cm-dish batch, with potential further reduction postpurification steps.
Hence, the establishment and evaluation of production systems and purification methods that can be effectively scaled in academia is imperative, as novel gene therapies are developed and the field continues to advance. 27 –29 When examining transfection yields for LVs expressing diverse gene cassettes, it became apparent that longer coding sequences resulted in slightly lower titers, as anticipated. 19 Furthermore, HYPERFlask® contributed to better productivity due to a 10-fold higher cell yield that increases capacity and reduces processing time and incubator space. These multilayer flasks use a gas-permeable film to enable efficient gas exchange, contributing to better productivity and supporting the results here observed. 30
Just as there are entire companies focused on the rational design of viral capsids, an industry has sprung up around viral vector production. 10,18,31 –33 Inefficient manufacturing protocols can result in viral batches with low yields, shorter shelf life and impaired purity, hindering their therapeutic efficacy. 9,18 Currently, there is a shift toward using serum-free media in suspension culture processes, as there are no serum-derived contaminants to eliminate, resulting in significantly higher vector purity from the beginning of DPS. 34,35 While these can be scaled down to smaller shakers in a research laboratory, this type of manufacturing is not typically the gold standard platform in academia; instead, adherent cell cultures are more commonly used, requiring closer attention to DSP and purification methods.
The removal of host cell proteins and transgene contaminants from the viral pool has been a concern in the field for decades. It is known that the presence of therapeutic transgenes in the viral solution can significantly reduce viral titers, either by exerting cytotoxic effects on producer cells, disrupting their metabolism, or interfering with the assembly and functionality of the vector virions, ultimately affecting potency and effectiveness. 13,14 Maunder et al. previously attempted to implement a “Transgene Repression in Vector Production” (TRiP) system to block the translation of one or more transgenes during viral production, thereby inhibiting the release of proteins with cytotoxic or apoptotic effects on the system. 14 Similarly, Blahetek and colleagues explored the use of artificial microRNAs for the transient suppression of toxic transgenes during viral production, successfully achieving effective silencing of the transgene and enhancing vector yields. 36 More recently, Banos-Mateos et al. described several strategies to reduce retro-transduction during LV manufacturing by blocking specific receptors on the surface of producer cells, thereby preventing viral interaction and entry. 13 It is noteworthy that when transgenes are driven by inducible promoters rather than constitutive ones, expression is inherently suppressed until the activator is introduced into the system, 37 contributing to a reduction of transgene contaminants in the viral pool even in the presence of retro-transduction.
When such strategies are not feasible, however, the use of effective purification methods is imperative for the proper elimination of contaminants before quality control assays and transduction of target cells can proceed. This ensures that the final viral preparations meet the necessary standards for efficiency and safety. While previous studies have independently addressed transgene silencing and purification techniques—such as Kutner et al., who described common purification methods such as ion exchange chromatography and polyethylene glycol 6000 precipitation, 38 as well as a simplified LV production protocol using HYPERFlask® vessels, 39 or Kuroda et al., who introduced PEI-mediated transfections for more consistent lentiviral batch production 40 —our focus was on the intersection of these approaches. Specifically, we aimed to determine which commonly used purification methods are most effective in reducing secreted transgene background within the viral pool. One important aspect not addressed in this study, which could be explored in future research, is the percentage of producer cells that were actually retro-transduced compared with those that were only transiently transfected. This could be further investigated by analyzing transgene integration in producer cells at the gene level through quantitative PCR, using specific primers to quantify the target transgene, rather than solely measuring it at the protein level as done in the present study.
When evaluating the purification efficiency of various research-scale methods in reducing transgene background, TFF demonstrated superior performance compared to others. This approach is known to be highly scalable and easily adapted for large-scale productions, being suitable for both laboratory-scale research and industrial manufacturing. 41,42 Furthermore, by allowing for gentle filtration of the sample through a semi-permeable membrane, TFF minimizes product loss, as reflected by the lowest % decrease in viral titrations. Moreover, TFF was effective in removing transgene background contaminants, resulting in batches with the lowest levels of GLuc. TFF is also known to be effective in removing contaminants such as aggregates, host cell proteins, and DNA from samples via size-based separation, resulting in higher purity of the final product 43 compared with density-gradient purification techniques, for example, that may not provide as effective removal of contaminants. 44,45 These can directly affect the health and viability of cells that are treated with nonpurified viral batches, which can also be a potential reason for decreased expression and lower transduction efficiency observed in those batches (Fig. 5). Notably, similar MOIs resulted in different transduction efficiencies in nonpurified viral batches (Figs. 4 and 5). This suggests that the different viral manufacturing scales—10 cm and HYPERFlask®, respectively—yield varying numbers of viable, fully mature viral vectors, indicating that manufacturing scale can affect viral integrity, which was not assessed in these studies. This issue could potentially be minimized through size-exclusion chromatography, which can effectively remove empty viral particles. 46
Although highly efficient, TFF can also present some challenges, such as shear sensitivity of some biomolecules to the shear forces generated during the process, which can result in potential product degradation; 47 membrane fouling due to accumulation of residues on the membrane surface, leading to reduced filtration efficiency and increased operating costs; limited molecular weight cutoff range due to TFF membrane size limitations. 48 For viral applications, however, TFF is highly suitable and offers exceptional versatility, which is particularly essential in academic settings. Our assessments further demonstrated that viral integrity was preserved postpurification, with an observed enhancement in viral potency during transduction (Fig. 5), suggesting an efficient separation of genome-devoid (empty) and mutant (incompletely assembled) virions from fully mature and functional vectors. 49
In conclusion, our study tackles a critical challenge in viral vector manufacturing: the efficient purification of viral pools and the reduction of secreted transgene background using readily available research methods. Moving forward, our report can contribute to paving the way for advancements in gene therapy research, 6 offering valuable insights for the DSP of viral vectors that encode secreted therapeutic transgenes.
Footnotes
ACKNOWLEDGMENTS
The authors thank Rick Cohen for sharing reagents such as PEI.
AUTHORS’ CONTRIBUTIONS
R.B.: Conceptualization, Investigation, Methodology, Data curation, Formal analysis, Visualization, Writing—original draft. A.B.: Conceptualization, Investigation, Methodology, Data curation, Formal Analysis, Visualization, M.T.: Investigation, Methodology, Data curation, B.P.: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing—review and editing.
AUTHOR DISCLOSURE STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FUNDING INFORMATION
The authors declare that financial support was received for the research and publication of this article. Financial support for this project was provided by the National Institutes of Health (NIH) (Grant 1R01EB028782).