
Abstract
Complement-mediated thrombotic microangiopathy (TMA) in the form of atypical hemolytic uremic syndrome (aHUS) has emerged as an immune complication of systemic adeno-associated virus (AAV) gene transfer that was unforeseen based on nonclinical studies. Understanding this phenomenon in the clinical setting has been limited by incomplete data and a lack of uniform diagnostic and reporting criteria. While apparently rare based on available information, AAV-associated TMA/aHUS can pose a substantial risk to patients including one published fatality. Reported cases were originally limited to pediatric Duchenne muscular dystrophy patients receiving micro- or mini-dystrophin transgenes via AAV9 but have subsequently been reported in both pediatric and adult patients across a range of disorders, transgenes, promoters, and AAV capsid types. This article provides an introduction to the complement system, TMA and aHUS, and anticomplement therapies, then presents clinical reviews of AAV-associated TMA/aHUS cases that have been reported publicly. Finally, exploration of risk factors and current and future mitigation approaches are discussed.
INTRODUCTION
In 2017, the U.S. Food and Drug Administration (FDA) announced the approval of Luxturna® (voretigene neparvovec-rzyl), “the first directly administered gene therapy approved in the United States that targets a disease caused by mutations in a specific gene.” 1 Since that milestone, adeno-associated virus (AAV)-mediated gene transfer has become an established treatment modality for a growing range of ophthalmological, hematological, and neurodevelopmental/neuromuscular disorders (Table 1).
FDA-approved AAV gene therapies
AAV, adeno-associated virus; FDA, U.S. Food and Drug Administration; IV, intravenously.
As clinical experience with AAV gene therapy has evolved, so has understanding of the associated risks, mostly immune-related, as well as benefits. Preexisting antibodies against AAV capsids and the resultant potential blunting of efficacy as well as enhanced secondary immune response have led most clinical development programs to exclude patients with total and/or neutralizing antibodies above a trial- and assay-specific threshold. Liver inflammation is a widely observed adverse event (AE) that has prompted use of immunosuppressant and immunomodulatory drugs either reactively or, more commonly, prophylactically, to mitigate the risk of immune-mediated liver toxicity.
A more recently emerging immunological safety issue in association with AAV gene therapy is complement-mediated thrombotic microangiopathy (TMA). This phenomenon had not been observed in preclinical studies so was unexpected when it first occurred in the clinical setting.
TMA is a class of disorders that share three canonical features: thrombocytopenia, hemolytic anemia, and end-organ damage. A specific type of TMA, atypical hemolytic uremic syndrome (aHUS), is defined by the triad of thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and acute renal failure. 2,3 Hemolytic uremic syndrome (HUS) that is not caused by bacterial toxins (typical HUS) is classified as “atypical” (aHUS) and is driven by complement activation. Reported cases of TMA associated with systemic AAV administration most closely resemble complement-mediated aHUS, although confirmatory evidence of complement activation has not always been provided. Because the nomenclature used for reported cases and in prescribing information has varied, this review will use the terms TMA or TMA/aHUS for simplicity. Additional information regarding TMA and aHUS is provided in a later section.
The index case of TMA associated with AAV gene therapy was reported in 2018 in Solid Biosciences’ systemic AAV9 gene therapy (SGT-001) program for Duchenne muscular dystrophy (DMD; NCT03368742). Here, the first participant, a nonambulatory adolescent boy, developed thrombocytopenia (8 days after dosing) followed by renal dysfunction and hemolytic anemia, the classic TMA triad. 4 Complement activation was confirmed biochemically. 5 TMA was subsequently observed in a subset of pediatric DMD patients receiving intravenous AAV9 micro- or mini-dystrophin, and complement-mediated TMA has since been reported with other AAV capsid types and patient populations across a variety of ages and background disease states. While posing a clinically significant risk when it occurs, it remains a rare phenomenon and the underlying factors driving individual cases remain unknown.
This article will present a brief overview of the complement system, TMA, and anticomplement therapies, then review the publicly reported cases of complement-mediated TMA associated with AAV gene therapy. Additional comments about potential risk factors and underlying mechanisms will be limited in scope and when present, focus on human data. The reader is directed to other recent reviews on complement activation and TMA/aHUS associated with AAV gene therapy for additional perspectives. 6 –8
An acknowledged limitation of this case review is the incompleteness and inaccessibility of potentially relevant information about these and other AAV safety events. This stems from a number of factors including sponsors’ different thresholds and criteria for public disclosure, materiality considerations for public companies, concerns around revealing proprietary information, and limitations of postmarketing safety data collection systems. The review closes with a call to action with respect to greater transparency and more widespread data sharing within the field of AAV gene therapy.
OVERVIEW OF THE COMPLEMENT SYSTEM AND TMA
The complement system is a branch of the innate immune system that is hard-wired and fully active from birth, in contrast to the adaptive immune system consisting of antibodies and cellular mediators of the immune response that learns and adapts over time. The complement system serves as a kind of “first responder” in defense against pathogens and consists of a series of regulated protein cleavage events that culminate in a final common pathway and formation of the C5b-9 membrane attack complex (MAC) that inserts into membranes leading to lysis of the target. This is very effective against infectious organisms but can be problematic when the complement system is inappropriately active, causing collateral damage to host tissues.
There are three main complement pathways: (1) the classical pathway that is activated by antibody–antigen complexes, (2) the lectin pathway that recognizes sugar molecules on the surface of bacteria and some viruses, and (3) the alternative pathway, which is constitutively “on” at a low level and amplifies the overall cascade when it encounters pathogens. Figure 1 shows the main components of the complement system. In addition, there is extensive crosstalk between the complement system and the coagulation cascade, other elements of the innate immune system, as well as the adaptive immune system (Fig. 1).

Overview of the complement system. The three complement pathways (classical, lectin, and alternative) converge at C3, which drives downstream activation of C5 culminating in the formation of the C5b-9 membrane attack complex. Approved complement-inhibiting therapies are shown in red. MBL, mannose-binding lectin; MASP, mannose-binding lectin-associated serine protease.
THROMBOTIC MICROANGIOPATHY
Inappropriate complement activation is associated with a wide range of disorders, but the events that have been seen with systemic AAV gene therapy have all been episodes of TMA.
TMA is characterized by abnormal thrombi or clots that develop primarily in the microvasculature. Pathophysiologically, the process starts with damage to the endothelium that in turn stimulates a cascade of events including activation and consumption of platelets and clot formation. Nets of clot material are formed across the lumen of the blood vessels and physically impede blood flow leading to red cell fragmentation. Microvascular injury and clot formation can also lead to end-organ damage, particularly in the kidney (Fig. 2).
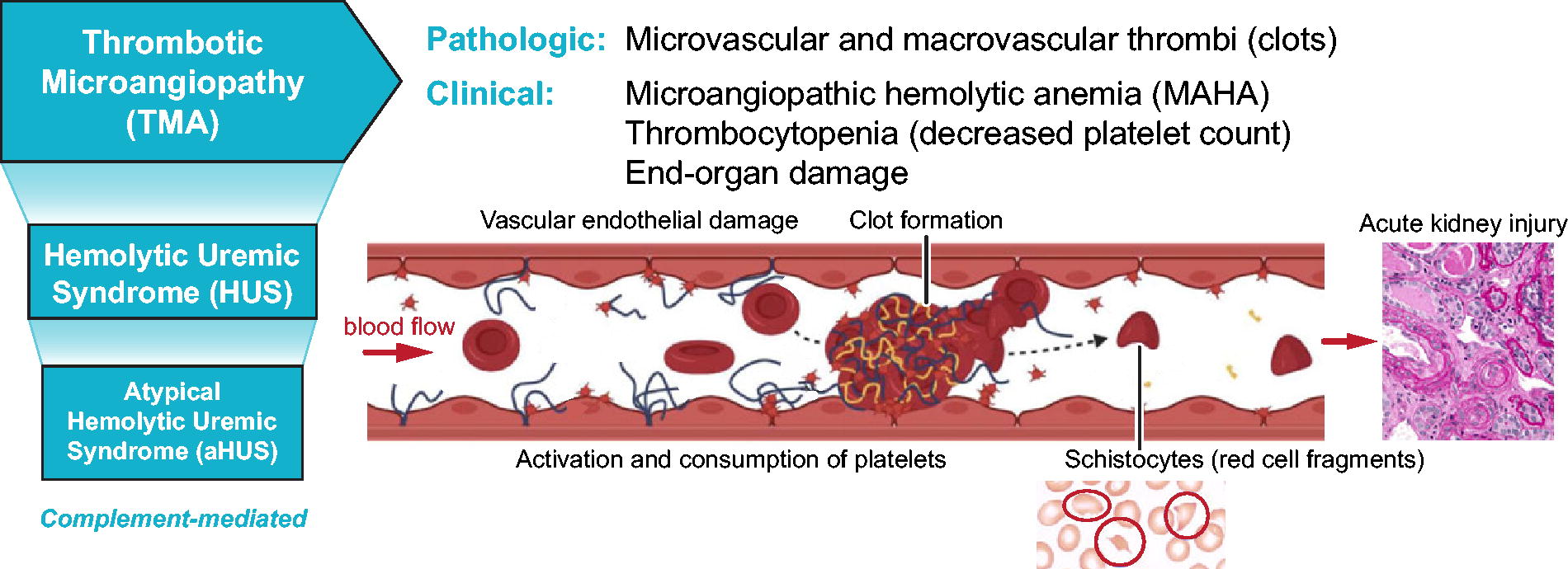
Thrombotic microangiopathy and atypical hemolytic-uremic syndrome (aHUS). aHUS is triggered by pathological complement activation leading to a cascade of endothelial damage, platelet activation, clot formation, and red blood cell fragmentation resulting in renal injury and dysfunction.
There are two main types of TMA that can have somewhat similar manifestations. One is thrombotic thrombocytopenic purpura that is caused by deficits in a thrombosis-regulating enzyme called a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13 (ADAMTS13). The other is HUS that is broken down into two main types. One (typical HUS) is caused by a bacterial toxin, Shiga toxin, that directly damages the endothelium. The other is aHUS that is caused by dysregulation of the complement system. There are many triggers leading to the pathological activation of the complement system in aHUS including genetic defects in inhibitory components of the complement cascade, malignancy, autoimmune diseases, and bacterial or viral infections. Cases of AAV-associated TMA most closely resemble the atypical form of HUS.
Clinically, aHUS is characterized by a classical triad of manifestations including thrombocytopenia, MAHA, and renal impairment. Other organ systems can also become involved including gastrointestinal, pulmonary, cardiac, the central nervous system, and skin.
ANTICOMPLEMENT THERAPIES
Several anticomplement therapies have been developed that target key steps in the complement pathway. C5 activation blockers such as eculizumab (Soliris® 9 Bkemv™10 ) and the next-generation C5 inhibitor ravulizumab (Ultomiris® 11 ) are monoclonal antibodies that bind to C5 and inhibit its cleavage and activation, thus interfering with assembly of the C5b-9 MAC at the end of the cascade. Upstream, recombinant or purified natural C1 esterase inhibitors such as Berinert® 12 and similar products help control complement activation by suppressing C1 activation. Finally, pegcetacoplan (EMPAVELI® 13 ), a polyethylene glycol (PEG)-conjugated pair of cyclic peptides, inhibits C3 and C3b, which sit at a critical juncture where the classical, lectin, and alternative pathways converge.
OVERVIEW OF TMA/aHUS CASES ASSOCIATED WITH AAV GENE THERAPY
Table 2 summarizes details about the vector, transgene, promoter, route of administration, dose level, indication, and population for publicly reported cases of TMA/aHUS associated with AAV gene therapy as of April 2024.
Publicly reported cases of TMA/aHUS associated with AAV gene therapy
Year and quarter are based on the initial case reported for each program. The age of the first affected patient is indicated in bold.
Q, quarter; AAV, adeno-associated virus; aHUS, atypical hemolytic uremic syndrome; TMA, thrombotic microangiopathy; DMD, Duchenne muscular dystrophy; vg, vector genomes; kg, kilogram; IV, intravenous; MCK, muscle creatine kinase; N/A, not applicable.
Initial cases: pediatric, intravenous AAV9
Intravenous AAV9 micro- and mini-dystrophin in boys with DMD
DMD is an X-linked disorder caused by loss-of-function mutations in DMD, the gene encoding the muscle protein dystrophin. Dystrophin deficiency causes progressive skeletal muscle degeneration and cardiomyopathy with loss of motor function and death in early adulthood. Gene transfer approaches utilize a condensed form of the dystrophin gene (micro- or mini-dystrophin) that can fit within the limited carrying capacity of AAV vectors (Table 3).
TMA cases in Solid Biosciences and Pfizer AAV9 programs for Duchenne muscular dystrophy
vg, vector genomes; kg, kilogram; S/S, signs/symptoms; DMD, Duchenne muscular dystrophy; yr, years; d, days; K, thousand; TMA, thrombotic microangiopathy; pt, patient; SAEs, serious adverse events.
The initial cases of what came to be understood as TMA/aHUS occurred in boys with DMD in Solid Biosciences’ SGT-001 AAV9 microdystrophin program. 4 The first case, a nonambulatory adolescent, developed rapid onset of thrombocytopenia followed by evidence of hemolytic anemia (schistocytes on peripheral blood smear) and renal impairment (elevated creatinine) approximately 1 week after gene therapy administration. Genetic testing for ADAMTS13 and complement factor mutations was negative. Activation of the complement system was confirmed biochemically. He received a course of two eculizumab doses with a third dose at the tail end of his recovery. The patient recovered completely without sequelae.
Subsequent study participants also developed TMA/aHUS; those in whom complement factors were measured displayed decreased C3 and C4 and elevated C5b-9. 28 Anticomplement therapy with eculizumab was implemented once evidence of complement activation was detected and one participant required both eculizumab and Berinert® (C1 esterase inhibitor). TMA occurred at both 5E13 and 2E14 vg/kg dose levels of SGT-001.
Cases were limited to the Solid Biosciences DMD program until Pfizer reported aHUS-like TMA in their fordadistrogene movaparvovec AAV9 mini-dystrophin program for DMD. Ultimately, a total of three participants developed TMA/aHUS that was treated with eculizumab along with platelet transfusion and temporary hemodialysis where needed. All three participants had received a dose of 2E14 vg/kg of the investigational gene therapy and a prophylactic glucocorticoid regimen. 5 Based on publicly available information, all DMD patients who experienced TMA/aHUS in the Solid Biosciences and Pfizer programs made a full recovery. 15 Complement-mediated TMA was not implicated in the first of two fatalities (a nonambulatory 16-year-old) reported in the Pfizer DMD gene therapy program. 5 The second fatality occurred in a study of younger DMD patients (2–<4 years old) and was still under investigation as of a June 2024 press release. 16
Onasemnogene abeparvovec-xioi (Zolgensma®) in infants and toddlers with spinal muscular atrophy
Spinal muscular atrophy (SMA) type 1 is an infantile-onset, rapidly progressive and fatal neuromuscular disorder caused by biallelic loss-of-function mutations in the SMN1 gene. Zolgensma® (onasemnogene abeparvovec-xioi [OA]), an AAV9-mediated gene therapy for SMA, was first approved in 2019 (Zolgensma prescribing information). 29 In mid-2021 a series of publications reported the first non-DMD cases of TMA/aHUS in SMA patients receiving OA (Table 4).
TMA cases in Novartis’ onasemnogene abeparvovec (Zolgensma®) program for spinal muscular atrophy
vg, vector genomes; kg, kilogram; S/S, signs/symptoms; K, thousand; SMA, spinal muscular atrophy; pt, patient; mo, months; yr, years; d, days; K, thousand; PO, per os (by mouth); PRBC, packed red blood cells; pt, patient; MAHA, microangiopathic hemolytic anemia; LFTs, liver function tests; LDH, lactate dehydrogenase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; TMA, thrombotic microangiopathy.
All these events occurred in the context of expanded access or postmarketing; TMA/aHUS had not been observed in the OA clinical development program. Review of these cases revealed some common themes regarding potential risk factors as well as onset and clinical course. TMA/aHUS was often preceded by infection, vaccination, or both within approximately 2 weeks prior to gene therapy administration. The first signs and symptoms emerged in a 5–8-day time frame after OA treatment, with classical TMA/aHUS manifestations and a range of renal signs including low urine output, hypertension, and blood and protein in the urine. Although nonspecific, nausea and vomiting were often the first clinical signs of emerging TMA/aHUS.
Patients received a variety of therapies including plasmapheresis, which is a traditional approach that was more widely used in the pre-C5 inhibitor era. Other treatments included glucocorticoids and eculizumab along with interventions addressing specific manifestations such as antihypertensives, diuretics and other fluid management measures, and platelet and red cell transfusions.
According to literature reports, all these SMA patients recovered fully with the exception of three who had persistent hypertension as of the time of the reports and one of whom had nephrotic syndrome that eventually resolved after 3 months. 17,18
Sadly, in July 2022, the first fatal case of TMA/aHUS was reported in a 6-month-old girl with SMA who had received OA at age 4 months via an early access program. 19 Her case occurred before the phenomenon of complement-mediated TMA/aHUS associated with AAV gene therapy had been published or widely recognized. The first clinical signs were noted on day 8 postdosing, with lethargy, vomiting, thrombocytopenia (nadir 22K), and elevated liver transaminases. She subsequently developed acute renal failure requiring hemodialysis, and hemolytic anemia. Treatment with eculizumab on day 13 led to recovery except for renal function. However, TMA recurred on day 19 likely triggered by hospital-acquired Staphylococcus epidermidis infection. Unfortunately, she succumbed on day 40 due to hypovolemia, sepsis, and cardiac dysfunction. Investigation of possible contributing factors revealed a variant of unknown significance in the gene encoding complement factor I, a regulatory protein that inactivates C3b and C4b.
First adult and non-AAV9 cases
AAV serotype C102 in an adult with Fabry disease
Fabry disease is an X-linked lipid storage disorder caused by mutations in the GLA gene encoding alpha-galactosidase-A (AGA) leading to substrate accumulation, particularly Gb3, and multiorgan pathology. In October 2021 4DMT reported a case of TMA/aHUS in a 32-year-old man who was the second of three initial participants dosed in their 4D-310 GLA gene therapy program for Fabry disease cardiomyopathy. 21 This was the first known case in an adult and the first associated with a non-AAV9 capsid, C102, a novel synthetic capsid selected for its cardiotropism. Laboratory findings included elevated creatinine (CTCAE Grade 3—severe) indicative of renal impairment along with thrombocytopenia (Grade 2—moderate), both noted on day 8 after gene therapy administration. This time course was consistent with that observed in other cases of AAV-associated TMA/aHUS with signs emerging approximately 1 week after AAV dosing. The participant was hospitalized for observation and supportive care and discharged after 4 days. He received no anticomplement treatment or hemodialysis. By day 15, renal function and platelet counts were improving, each with a decrease of 1 on the Common Terminology Criteria for Adverse Events (CTCAE) severity scale; the patient ultimately made a full recovery with no sequelae. This participant was notable for being the only one not receiving enzyme replacement therapy (ERT) and having the highest anti-AGA antibody titer (1:99,900) prior to dosing with 4D-310 consistent with the concept of antibody-driven activation of the classical complement pathway.
In January–February 2023, 4DMT announced additional cases of TMA/aHUS for a total of three across two clinical trials in the United States and Asia-Pacific region in which six participants had received 4D-310 at a dose of 1E13 vg/kg with corticosteroid immunomodulation. 22,23 All resolved within approximately 2–4 weeks. One of the events was Grade 4 (potentially life-threatening) involving a 69-year-old man with underlying kidney dysfunction who required temporary hemodialysis. Further investigation revealed he had preexisting activation of the alternative complement pathway prior to gene therapy administration. 4DMT voluntarily suspended further dosing and the FDA placed the program on clinical hold. In October 2023, 4DMT announced alignment with the FDA on a plan to lift the clinical hold based on addition of risk mitigation measures including predose testing for complement activation and immune prophylaxis consisting of rituximab and sirolimus to abrogate antibody responses that may drive complement activation and TMA. The 4DMT TMA cases and associated immune findings and mitigation have been reviewed in Salabarria et al. 2024. 28
AAV9 in an adult with Danon disease
Danon disease is an X-linked disorder caused by loss-of-function mutations in the LAMP2 gene encoding lysosome-associated membrane glycoprotein 2 (LAMP2). This protein is responsible for fusion of lysosomes and autophagosomes and LAMP2 deficiency leads to impaired autophagy. Danon disease is characterized clinically by hypertrophic cardiomyopathy as well as skeletal muscle pathology and often cognitive impairment.
In November 2021 Rocket Pharma reported a case of TMA/aHUS occurring in a 20-year-old man with Danon disease who was one of two high-dose participants administered Rocket’s RP-A501 AAV9 LAMP2B vector at a dose of 1.1E14 vg/kg intravenously (IV). Both participants received immune prophylaxis consisting of rituximab, tacrolimus, and prednisone. The affected participant developed Grade 4 (severe) TMA with evidence of complement activation along with Grade 4 thrombocytopenia and acute renal failure that required temporary hemodialysis. The episode fully resolved with eculizumab and supportive treatment with return to baseline kidney function within 2–3 weeks. This participant was reported to have had preexisting anti-AAV immunity and was the heavier of the two and therefore exposed to a higher total vector load. 24
Based on this event, FDA placed the RP-A501 program on clinical hold in May 2021. The hold was lifted in August 2021 based on new risk mitigation measures including discontinuing further dosing of adolescents/adults in the 1.1E14 vg/kg cohort and modifying the immune prophylaxis regimen. The total daily corticosteroid dose was reduced to lessen the risk of steroid myopathy, a known vulnerability in Danon disease patients that had occurred in the high-dose group, and tacrolimus was replaced with sirolimus to lower potential renal liability. In addition, frequent monitoring for early signs of TMA was implemented.
The subsequent low-dose pediatric cohort consisted of two 12-year-old participants who received RP-A501 at a dose of 6.7E13 vg/kg along with the modified immunomodulatory regimen. A program update in September 2023 reported normal platelet counts, minimal complement activation, and no complement-associated AEs. 30 Review of these two children and their immunomodulatory regimen is provided in Salabarria et al. 2024. 28
AAV serotype LK03 in children with methylmalonic acidemia
Methylmalonic acidemia (MMA) is an organic acidemia characterized by impaired metabolism of methylmalonic acid. The most common form is caused by autosomal recessive mutations in the MMUT gene leading to deficits in methylmalonyl-CoA mutase activity.
The second report of TMA associated with a non-AAV9 capsid came from LogicBio’s gene editing program for children with MMA caused by MMUT mutations. 25 Their approach used a novel synthetic capsid, LK03, that is similar to wild-type AAV3B to deliver a copy of the human methylmalonyl-CoA mutase gene that is modified to permit genomic insertion at the albumin locus. Patients received an IV infusion of vector at a dose of 5E13 vg/kg. 31
No cases of TMA were observed in children 3–12 years of age. However, in October 2021 the company reported a patient in the younger 6-month–2-year-old group who displayed signs of TMA at the 2-week visit, including hemolytic anemia and acute kidney injury. No thrombocytopenia was observed, but based on what has been seen with other cases, a clinically silent drop in platelet count can develop and then subsequently resolve all within the first 2 weeks posttreatment; therefore, a transient thrombocytopenia may not have been detected. In keeping with the theme of recent infection and/or vaccination, this patient had an antecedent viral infection that delayed gene therapy dosing by a few days. The patient was managed supportively in the hospital and recovered. This serious AE prompted changes to the protocol including revised eligibility criteria and increased monitoring but no modification to the immunosuppressive regimen.
In February 2022 the company reported a second patient in the younger age cohort who developed the classical aHUS triad of decreased platelets, anemia, and renal dysfunction less than 1 week after dosing. The patient was hospitalized and treated with IV fluids, eculizumab, red cell and platelet transfusions, and hemodialysis. Both affected children recovered fully within a matter of weeks.
Based on these two cases, the FDA imposed a clinical hold in February 2022 that was lifted in May 2022, with risk mitigation measures including frequent testing for complement activation and use of a complement inhibitor when laboratory findings of TMA were seen. 26,27 No further patients were dosed and as of April 2023 the program was reported as “terminated due to low likelihood of clinical benefit in treated participants.” 31
TMA prevalence
Accurate estimates of the prevalence of TMA overall and in different AAV gene therapy programs are limited by multiple factors, including a lack of uniform diagnostic and reporting criteria, different thresholds for public disclosure, and unclear denominators for calculation of rates. In addition, voluntary safety data collection systems such as registries may be inconsistent, incomplete, or duplicative and do not always include or describe standards for how cases are discerned or categorized. Strengthened expectations around safety reporting, development of consensus diagnostic criteria, harmonization of data formats, and incentivizing data sharing will enhance understanding of the scope of the problem as well as risk factors and mitigation strategies.
A specific consideration in case discernment efforts for TMA is the distinction between isolated thrombocytopenia and full-blown TMA/aHUS. Self-limited thrombocytopenia can be observed within the first 2 weeks after systemic AAV gene transfer. 28,32 It is noted as a distinct entity in the Warnings and Precautions section of the Zolgensma prescribing information and may be mechanistically distinct from TMA/aHUS. 29,33 In contrast, AAV-associated TMA, although serious and potentially life-threatening, is apparently a rarer phenomenon. Among children treated with Zolgensma who constitute the largest population to have received systemic AAV9, out of more than 4,000 patients dosed in clinical trials, managed access, and commercial settings as of October 2024, a total of 37 cases of TMA and 180 cases of thrombocytopenia had been reported as of September 30, 2024 according to the voluntary (and hence incomplete) FDA Adverse Event Reporting System (FAERS) (personal communication; Novartis medical information, November 4, 2024). This corresponds to an estimated TMA prevalence of 0.9% and is consistent with findings from the RESTORE registry (n = 167) in which one case of TMA was reported (0.6%) and a global managed access program with 1 TMA case among 102 patients ≥8.5 kg (0.9%). By comparison, the rate of isolated thrombocytopenia in RESTORE was 13.8%. 34 In a smaller case series (n = 8), all patients demonstrated a posttreatment drop in platelet counts, of which 75% qualified as thrombocytopenia. Among these participants, the platelet nadir occurred between 6 and 8 days posttreatment. 32 There were no cases of TMA.
In the Zolgensma example, a potential source of uncertainty in estimating TMA/aHUS prevalence involves the label’s recommendations regarding postdosing monitoring. No laboratory evaluations are explicitly specified until week 1 and both the timing and extent of laboratory testing do not cover critical, early-emerging TMA markers. This, coupled with undisclosed or inconsistently defined and applied diagnostic criteria for TMA/aHUS, may contribute to under- or misreporting.
Underlying factors
It is not known why some individuals develop TMA or why some AAV gene therapy programs have had a higher proportion of TMA cases than others. It is likely that multiple factors combine to create an overall predisposition for a given patient.
AAV serotype was initially implicated as a common element since the first cases in DMD and SMA (along with the later Danon disease program), all utilized AAV9. Additional cases revealed that TMA could also occur with novel synthetic capsids that are not related to AAV9 (clade F) such as C102, a modified version of AAV2 (clade B), and LK03 that is most akin to AAV3b (clade C). 28,35,36 Nevertheless, the vast majority of known cases have occurred with high-dose systemic AAV9, a point worthy of heightened awareness among clinical users of AAV9-based therapies. The preponderance of AAV9 among reported TMA cases but no such association with other capsids such as AAV8 and AAVrh74 administered IV at similar or higher doses in the same disease background (DMD) is a distinction that invites comparative data analysis. While yet to be elucidated, capsid characteristics are likely to play an important role in that the capsid surface is the first point of contact with the host immune system as well as with the endothelial milieu where TMA begins.
The one universal commonality to date is IV administration, which, as just mentioned, is consistent with the role of endothelial insult in the pathogenesis of TMA. Related potential factors include total viral load and product quality attributes, the ratio of empty to full capsids (empty-full ratio), “partials” with packaging of truncated vector genomes, or exposed capsid-associated or free DNA that can be highly immunogenic. True like-for-like comparisons across products based on these parameters are hampered by differences in titering and other analytic methods as well as assay performance and quality specifications, all of which are usually proprietary.
Among early TMA/aHUS cases there was a pattern of greater risk at higher nominal AAV doses (5E13–2E14 vg/kg) and heavier patients, with the hypothesis that there is a greater likelihood of inducing viral-mediated pathological processes with greater total viral exposure. In some cases, setting an upper limit on participant weight was implemented to mitigate this factor. Along similar lines, an unfavorable empty-full ratio with additional load from immunogenic but nonefficacious viral particles (i.e., empty capsids) was considered a contributor, prompting improvements in manufacturing processes to reduce risk. 14
The immune milieu in a given patient is likely an additional determinant. Predisposing conditions may include the presence of anti-capsid antibodies or antibodies against the transgene product (adult Fabry and Danon patients), preexisting complement activation (senior Fabry patient), antecedent infection or vaccination (Zolgensma-treated and MMA children), or a genetic defect in complement regulatory factors (underlies many cases of non-AAV aHUS; possible factor in fatal OA case). 19
Overall, as more cases have been reported, it has been difficult to develop a unified picture regarding the underlying mechanisms and risk factors. To date, no common elements have emerged in terms of capsid type, promoter or transgene, or with patient age or size (Table 1).
A perhaps underrecognized factor is background disease state.
Duchenne muscular dystrophy
The first cases of AAV-associated TMA occurred in DMD patients. In addition to the high doses of systemic AAV9 needed to reach and transduce muscle (5E13–2E14 vg/kg), dystrophic muscle itself is in a proinflammatory state so may be primed for immune activation by a high viral load. 37 Immunohistochemistry of DMD muscle biopsies has shown muscle-specific expression of C5b-9 indicative of local complement activation. 38 Proteomic analysis of circulating biomarkers in glucocorticoid-naive DMD patients showed decreased levels of two complement inhibitors, complement decay accelerating factor and CD59, which increased with glucocorticoid treatment. 39
Spinal muscular atrophy
Complement has been implicated in some pathological mechanisms occurring in SMA. In SMA mice, C1q and C3 (classical complement pathway) are upregulated and involved in increased elimination of proprioceptive synapses associated with survival motor neuron (SMN) deficiency. 40 In addition, SMN deficiency is associated with abnormalities in microvascular development, endothelial repair, and platelet function. 41,42
Both animal models and SMA patients display defects in the development and maturation of retinal blood vessels. 41 Reduced angiogenesis is also seen in SMN-deficient cultured human umbilical vein endothelial cells. 41 In addition, levels of circulating endothelial cells, a biomarker of endothelial damage, as well as decreased endothelial progenitor cells in SMA patients are indicative of an imbalance between endothelial injury and repair. 41 SMN protein is expressed in platelets, and SMA model mice have abnormally elevated production of megakaryocytes and platelets, as well as circulating microclots and peripheral vascular dysfunction and occlusion. 41 Coagulation and platelet abnormalities are a common finding in SMA patients. 44 Taken together, these findings support complement dysregulation and intrinsic platelet, endothelial, and small-vessel pathology, all predisposing factors for TMA, as fundamental features of the SMA phenotype. 41,44
Fabry disease
Fabry disease is associated with abnormal C3-mediated complement activation including C3 deposition in the kidney, which may compound the damage caused by Gb3 accumulation. 45 Plasma proteomic analysis has demonstrated elevated iC3b and C4B, both effectors in the complement cascade, in treatment-naive adults with Fabry disease compared with age- and sex-matched controls. 43,45 Levels of iC3b (and C4B to a lesser extent) decreased after ERT with agalsidase beta (Fabrazyme®); levels rose transiently along with substrate levels during suspension of ERT during a manufacturing-related shortage. 45 ERT-associated changes in complement factor levels were also reported in pediatric patients as measured before and after 26 weeks of agalsidase alfa treatment. These findings support an association between alpha-galactosidase deficiency and perturbations in the complement system as an underlying pathological mechanism in Fabry disease.
Danon disease
Danon disease, unlike other vacuolar myopathies such as X-linked myopathy with excessive autophagy and others, is not associated with complement deposition on membranes of affected muscle cells. 46 Endothelial abnormalities were identified in a mouse model of Danon disease but those findings were not replicated in a study of small-vessel pathology in adults with LAMP2 deficiency. Aside from a general connection between deficient autophagy and endothelial pathology, 47 there is no direct evidence of complement activation or an endotheliopathy associated with Danon disease.
Methylmalonic acidemia
One form of MMA, the rarer Cb1B type with homocystinuria, is a disorder of vitamin B12 metabolism and is known to be associated with HUS in pediatric patients (including infants) as well as late-onset adults. 48 –50 However, the two pediatric cases of AAV-associated TMA involved only patients with isolated MMA caused by pathogenic MMUT mutations, which has not been reported to be associated with aHUS.
MITIGATION AND FUTURE DIRECTIONS
Because of its rarity and the diversity of programs and patient populations in which it has occurred, AAV-associated TMA has eluded characterization with respect to predisposing factors. That said, given the possible contribution of a variant of unknown significance in a complement inhibitory factor in the published OA TMA fatality, 19 genetic testing for abnormalities in the complement pathway may be worth considering.
In terms of best practices for TMA/aHUS mitigation and management, some common themes have emerged as clinical experience continues to accrue. The following information is not intended as medical advice but rather an overview of some trends and recommendations from product prescribing information, peer-reviewed publications, and expert consultation. These include avoiding vaccination in the 2 weeks prior to AAV administration 51 (some clinical trials require 4 weeks) and waiting to dose until any active infection is resolved 29 (a waiting period of at least 2 weeks is recommended in published guidelines 51 and stipulated in some clinical trial protocols).
After AAV administration, frequent laboratory surveillance is recommended, particularly in the first 2–4 weeks after AAV administration, 28,29,51 including platelet counts, red blood cell morphology (manual peripheral smear to detect schistocytes), and complement testing (comprehensive panel, or if necessary, single tests to detect decreased C4 and particularly increased C5b-9). 28,29 D-dimer has also been mentioned as a helpful early marker of endothelial activation. 28 Creatinine can be an informative marker of renal status although interpretation needs to consider the background disease state; in conditions in which baseline creatinine levels are low (e.g., DMD), an increase to a level below the lower limit of normal may still reflect deteriorating renal function.
Approaches to post-AAV safety surveillance may also need to be tailored to the age of the patient population as the nature of the immune response along with clinical manifestations of TMA/aHUS may differ in infants, older children, and adults. 52
From a feasibility standpoint, real-time surveillance and detailed characterization of the complement response has been challenged by the paucity of qualified laboratories and long turnaround times for complement assays (particularly sC5b-9). In addition, the frequency and extent of testing for all safety parameters postdosing is constrained by blood volume limitations in the pediatric setting where most of the cases have occurred. Broader access to real-time complement testing (particularly sC5b-9) to guide clinical decision-making as well as more widespread availability of lower blood volume options for routine safety assessments in babies and children would be very helpful advancements for the field.
Despite the central role of complement activation in AAV-related TMA/aHUS, available information indicates that prophylactic use of complement inhibitors such as a C1 esterase inhibitor and/or eculizumab has not been successful in preventing new cases. 5 In addition, in gene therapy and other settings, concerns have been raised about preemptive use of eculizumab without evidence of high TMA liability because of the risk of serious meningococcal infection associated with C5 inhibitors.
A recent analysis of immune prophylaxis regimens, immune markers, and clinical outcomes in pediatric and adult participants from different AAV9 gene therapy programs has suggested that immunomodulation with rituximab (B cell cytotoxicity) and sirolimus (inhibition of B and T cell activation) can be effective in decreasing antibody production and preventing potentially deleterious immune responses. 28 No patients receiving this regimen developed complement activation or TMA/aHUS. These findings are the first published evidence supporting a specific immune prophylaxis regimen for AAV gene therapy; additional such studies are encouraged to further advance the field.
As with any intervention, benefit–risk must be carefully weighed when considering implementation of a preventive immune regimen. This is difficult in a setting where data are few and prognostic factors are unknown. The safety liabilities around complement inhibitors are well recognized. Both rituximab and sirolimus are highly efficacious but can have prolonged immunosuppressive effects lasting, in some cases, 6 months or more after discontinuation. 53,54 Identification and implementation of other preventive measures await further investigation.
The long-term success of AAV gene therapy depends on a comprehensive understanding of its safety profile as well as prognostic risk factors. This understanding can be deepened and accelerated by data sharing and pooled analyses of safety results from different AAV programs, including more subtle evidence of subclinical or “forme fruste” aHUS/TMA. In the DMD gene therapy space, publication of case studies in journals and safety-focused scientific meetings involving open sharing across advocacy, academic, and industry stakeholders have supported this aim. 55,56 The emergence of unexpected cases of skeletal, respiratory, and bulbar myositis and myocarditis in multiple AAV programs for DMD prompted a collaborative effort among academic experts and sponsors that resulted in the implementation of enhanced safety measures across programs. 55 Building on this success, larger-scale, therapeutic area-agnostic efforts are underway involving public, private, and academic partners to tackle the most pressing safety issues facing the field of AAV gene therapy (June 2023 NIH Workshop to Address Emerging AAV-Related Toxicities and four follow-on working groups: Anti-transgene Immune Rejection, TMA, Cardiac Adverse Events, and Hepatopathies). Practical implementation of safety data sharing and pooled analyses in an impartial and sponsor-deidentified setting is also being explored, with the FDA-funded Critical Path Institute initiative RDCA-DAP (Rare Disease Cures Accelerator-Data and Analytics Platform) emerging as a promising potential option. 57
Footnotes
ACKNOWLEDGMENTS
Many thanks to Daniel Levy and Richard Finkel for their insights regarding safety findings associated with systemic AAV gene therapy; Carsten Bönnemann and Francesco Muntoni for creation of a precompetitive forum for discussion of AAV safety among sponsors, academic, and public partners; Adam Shaywitz for his critical review during article preparation; and Michael Guerrero for his insights regarding AAV manufacturing and analytic methods. Medical writing support from Paginae Incorporated is gratefully acknowledged. Special thanks go out to all the patients and families who have participated in AAV gene therapy clinical trials.
AUTHOR’S CONTRIBUTIONS
G.L.: Conceptualization, data curation, writing—original draft preparation, and writing-reviewing and editing.
AUTHOR DISCLOSURE
As of the date of article submission, G.L. was a full-time employee of Aspa Therapeutics/BridgeBio Gene Therapy and a stockholder in BridgeBio Pharma. No conflicts of interest are applicable to this review article, which represents the author’s personal perspectives. The statements in this review do not necessarily reflect the views of Aspa Therapeutics/BridgeBio Gene Therapy and are not to be construed as medical advice.
FUNDING INFORMATION
No external sources of funding were used to support preparation of this review.