
Abstract
Adeno-associated virus (AAV)–associated gene therapy has been increasingly promising, in light of the drugs progressed to clinical trials or approved for medications internationally. Therefore, scalable and efficient production of recombinant AAV is pivotal for advancing gene therapy. Traditional methods, such as the triple-plasmid transfection of human embryonic kidney 293 cells in suspension culture, have been widely employed but often hampered by low unit yield. In this study, we optimized the cell culture process with high cell density up to 2 × 107 cells/mL by employing a perfusion culture system with centrifugation and medium exchange in shake flasks and perfusion device in bioreactor. Furthermore, we utilized a design of experiments strategy to systematically modulate a series of transfection-related variables including the quantity of plasmid DNA, the DNA-to-polyethylenimine ratio, incubation duration, and the impact of post-transfection feeding strategies on the yield of recombinant AAV (rAAV). Our comprehensive analysis and subsequent optimizations actualized a remarkable unit yield reaching nearly 2 × 1012 vector genomes (vg)/mL. Importantly, the resulting single-cell yield and biological activity were found to be comparable with those obtained from fed-batch cultures, underscoring the efficacy of our approach. Based on these findings, we investigated rAAV yield via high-density suspend culture in bioreactor, particularly focusing on cell aggregation and the use of perfusion technology. Intriguingly, we attempted to elevate the yield of an oversized recombinant coagulation factor VIII AAV843 vector by 3.5-fold, reaching a yield of 1 × 1012 vg/mL. Concurrently, the medium usage rate was only double that of batch feeding, thereby significantly shrinking the upstream cost of rAAV manufacture. In summary, this strategy significantly benefits large-scale AAV production for both commercial and clinical applications.
INTRODUCTION
Recombinant adeno-associated virus (rAAV) is a prominent gene delivery vector, favored for its exceptional gene transfer efficacy, precise tissue tropism, and low immunogenic profile. 1,2 This vector system is extensively applied in clinical trials to treat a broad spectrum of indications including rare genetic conditions, diseases of the central nervous system, ocular disorders, and muscular diseases. 3 Broadened indications highlight the stringent demand of large-scale AAV production. For instance, the therapeutic dosage for hemophilia A via systemic administration is projected to be 3 × 1013 vg/kg of body weight, 4 whereas the dosage for the treatment of Duchenne muscular dystrophy with a specific drug is 1 × 1016 vg/patient. 5,6 Further elaboration of rAAV production methodologies is imperative to ensure a sustainable and robust supply chain for rAAV-based therapies, thereby facilitating the advancement of gene therapy as a transformative medical modality. The classical method for producing rAAV involves the co-transfection of human embryonic kidney 293 (HEK293) cells with three plasmids: pGOI (Gene of Intresting) plasmid containing the target gene, pRepCap plasmid encoding the AAV replication-controlling rep gene and the capsid protein-expressing cap gene, and pHelper plasmid assisting AAV expression within the cell. 7 Large-scale suspension culture and transfection processes for HEK293 cells have been developed recently. 8,9 To maximize viral yield, transfection is typically performed during the logarithmic growth phase of the cells, when intracellular metabolic levels such as DNA replication and protein synthesis are high, providing favorable conditions for virus proliferation. However, there are some limitations of the composition in the culture medium during batch culture, the cell density in the mid-logarithmic growth phase is generally within 1–5 × 106 cells/mL. Under a certain single-cell yield, the total virus yield per unit volume is limited. Feeding and perfusion culture are common techniques used to increase cell density and ensure nutrient supply in the culture medium to maintain single-cell yield. Dozens of studies focused on such techniques to increase cell density for the production of recombinant viruses. Currently, the main equipment used in perfusion culture includes ATF/TFDF devices (Repligen) based on fluid pressure separation and Biosep devices (Applikon) based on acoustic precipitation separation. For instance, Hock and colleagues increased the HEK293 cell density to more than 7 × 107 cells/mL through ATF/TFDF perfusion culture, and Chahal’s group tried 2.5 × 107 cells/mL using perfusion culture, increasing the unit volume yield of Modified vaccinia Ankara (MVA) and influenza A virus by 7-fold. 9,10
High-density transfection requires ensuring not only cell viability during transfection but also transfection efficiency. The mixing of cationic polymer polyethylenimine (PEI) with plasmids under certain conditions for plasmid transfection into cells is an effective method for large-scale transfection. Backliwal and colleagues found that adding plasmid DNA and PEI directly to the cell culture medium can achieve high transfection efficiency at higher cell densities to produce recombinant proteins. 11 Nevertheless, the bottleneck of transfection efficiency at high densities remains unsolved. The composition of the culture medium, including anti-cell aggregation agents, metal ions, and nutrient provision, could significantly impact transfection efficiency and virus production. 12,13 A study used ATF and TFDF perfusion culture to increase the density of HEK293 cells to 5 × 106 cells/mL for triplasmid transfection to produce rAAV and studied post-transfection perfusion processes, achieving a 2-fold increase in yield compared with batch culture, reaching 6 × 1011 vg/mL. 14 CEVEC Pharmaceuticals constructed a Tet-induced stable cell line for the production of rAAV and increased cell density through an ATF perfusion culture system, achieving an rAAV yield of 1 × 1012 vg/mL. 15
In this study, we simulated perfusion culture by centrifugally replacing the medium in flasks to increase the density of HEK293 cells to 2 × 107 cells/mL, determining the logarithmic growth phase based on the doubling time. Through design of experiments (DOE), we meticulously evaluated various parameters, including plasmid concentration, DNA:PEI ratio, and incubation duration, with the rAAV yield as the pivotal response variable. During the post-transfection virus proliferation stage, the effects of perfusion culture and feeding culture on virus yield were studied. It was found that replacing continuous perfusion with feeding significantly increased virus yield, maintaining single-cell productivity and upregulating the yield of different serotype rAAVs by 3–5 times, with a maximum of 1.7 × 1012 vg/mL achieved. To further investigate the scale-up production process of perfusion culture, high-density culture of HEK293 cells based on perfusion was performed in a 3 L stirred bioreactor using Biosep, with optimized cell-specific perfusion rates (CSPR). With ameliorated transfection and culture conditions, the yield of an oversized (∼5.2 kb) recombinant FVIII AAV843, produced in high density (2 × 107 cells/mL), was increased by more than 3 times compared with fed-batch culture, reaching 9 × 1011 vg/mL. Simultaneously, the consumption of basal medium was only twice that of batch feeding culture, saving medium consumption per viral genome by 35%. These studies provide a theoretical basis for the further scale-up of commercial high-density production of rAAV and effective references for improving the efficiency of rAAV production.
MATERIALS AND METHODS
Cell flask culture
The HEK293 cells (CRL-1573, ATCC) screened by our laboratory were adapted for suspension culture and passage in shake flasks named HEK293-S03. A 125 mL triangular flask (Corning) with a culture volume of 15 mL was used, and the culture medium (Gibco) consisted of proprietary chemically defined basal and feed medium (Cytiva) supplemented with 8 mM glutamine (Merck-Sigma). The CO2 incubator shaker (Kuhner) was set at 37°C, 5% CO2, 80% humidity, and a rotation speed of 130 rpm with an orbital diameter of 50 mm. Cells were passaged at an initial density of 0.6 × 106 cells/mL and cultured until the density that reached 4–5 × 106 cells/mL. Medium exchange was performed when the cell density reached the passage density (5 × 106 cells/mL) by centrifuging at 300 g for 5 min and then replacing the culture medium with fresh medium containing additional 4 g/L glucose (Merck-Sigma) and 12 mM glutamine to the original volume for continued culture. The medium exchange on the day of transfection was completed before transfection.
rAAV triple plasmid transfection
The plasmids used for triple plasmid transfection were pHelper plasmid using Phelper (GenBank: AF369965.1), pRepCap using AAV serotypes 5/8/9 and AAV843 (Patent No. ZL201980053196.1), independently developed by our laboratory, pGOI containing the expression of CMV promoter-eGFP (ssDNA-ITR, 1.3 kb), CB promoter-coagulation factor VIII (ssDNA-ITR, 5.1 kb), and CB promoter-coagulation factor IX (dsDNA-ITR, 2.7 kb). All plasmids were provided by BeliefBioMed Pharmaceutical. The mixing ratio of the three plasmids pGOI, pRepCap, and pHelper was 1:2:2(w/w/w). PEI MAX (Polysciences) was used as the transfection reagent and prepared into a 1 mg/mL solution using PBS. DMEM (Thermo-Fisher Scientific) was used as the diluent, and the mixture was added to the cell suspension after the incubation time at room temperature. At 24 h and 48 h after transfection, a medium exchange to simulate perfusion was performed by centrifuging and replacing it with fresh medium to the original volume.
Experimental of design for high-density transfection conditions
Based on experience, initial parameters for high-density (about 2 × 107 cells/mL) transfection were set as follows: 0.5 µg of plasmid per 1 × 106 cells, DNA:PEI (w/w) ratio of 1:2, mixing volume of 15% of the total cell suspension volume, and incubation at room temperature for 4 min. A central composite design was used, and the experimental design table was created using JMP software (see Supplementary Table S1). The factors included plasmid dosage, DNA:PEI ratio, and incubation time. The ranges of these factors were:7.5–15 mg/L for plasmid dosage, 1:2–1:4 for DNA:PEI ratio, and 2–8 min for incubation time after mixing. The total mixing volume was fixed at 15%. The response variables were the rAAV genome titer and virus particle count. The results of the experimental design were analyzed using the least squares method in JMP software. For feed-batch transfection, the density was set at 5 × 106cells/mL, with 0.5 µg of plasmid per 1 × 106 cells, DNA:PEI (w/w) ratio of 1:2, mixing volume of 10% of the total cell suspension volume, and incubation at room temperature for 9 min.
Experimental design for feed addition after high-density transfection
The experimental design for the proportion of medium exchange (simulated perfusion rate) and feed addition after transfection is detailed in Supplementary Table S1. Fresh medium containing additional 2 g/L glucose and 12 mM glutamine was used for medium exchange, with a range of 0 − 200%. The feed used was the laboratory-screened feed A/B, mixed in a ratio of 10:1 with the culture medium, and the feed volume ranged from 0% to 15% of the culture volume. The feed addition was performed at 3 h, 24 h, and 48 h after transfection. Additionally, during each feeding, a final concentration of 14 mM glutamine was supplemented.
Bioreactor perfusion culture
The bioreactor used was the EZ-control 3 L stirred bioreactor (Applikon). A pitched blade impeller was used for stirring, and the bottom aeration was provided through pores with a diameter of 0.1 mm. The stirring speed ranged from 200 to 400 rpm. The dissolved oxygen was controlled at 40% through PID control of pure oxygen, and the pH was maintained at 7.1 ± 0.05 using CO2 and 8% sodium bicarbonate for adjustment. The cell separation equipment used for perfusion culture was the Biosep 10 L (Applikon), with separation parameters set at 2 W power, 30 s run time, and 10 min stop time. The perfusion inflow and outflow volumes were manually controlled daily, calculated using the formula V = (v1+v2) / 2 × CSPR / 24, where V is the hourly inflow and outflow rate, v1 is the current cell density (perfusion starts when cell density reaches 5 × 106 cells/mL), and v2 is the estimated cell density after 24 h based on the normal cell doubling rate (typically, v1 is 5 × 106 cells/mL on the first day, and v2 is 1 × 107 cell/mL after 24 h of perfusion; on the second day, v1 is 1 × 107cells/mL, and v2 is 2 × 107cell/mL), with V representing the culture volume.
Cell lysis and clarification process
The cell harvest solution was lysed for 30 min with 50 mM Tris (Merck-Sigma) buffer containing 0.1% Tween 80 (Merck-Sigma). Then, 5 U/mL benzonase (Merck-Sigma) and 2 mM MgCl2 (Merck-Sigma) were added, and the mixture was incubated at 37°C for 30 min. For high-density samples, the lysis process was performed after diluting the sample 4 times with PBS. The treated cell harvest solution from the process reactor was then clarified using a 3M depth filter.
Cell density and transfection efficiency detection
Cell density counting and GFP transfection efficiency determination were performed using the Cellex cell counter (Nexcelom). Cell viability was assayed using trypan blue staining according to the instrument’s instructions. Before testing, cells were digested with trypsin (Gibco) mixed 1:1 at 37°C for 5 min.
Metabolomics analysis
Cell culture medium was centrifuged at 300 g for 5 min, and the supernatant was collected. The concentrations of glucose, lactate, glutamine, and ammonium ions were measured using the Cedex bio (Roche Diagnostic) according to the instrument’s instructions.
qPCR quantification of rAAV genome titer
Sample preprocessing: For viral DNA release, 0.1% SDS was added to the cell lysate and incubated at 95°C for 1 h. qPCR was performed using primers (F-GCCTCAGTGAGCGAGC/R-AGGAACCCCTAGTGATGG) to detect the ITR sequence of rAAV, and the genome titer was determined using an external standard method. The standard curve was generated using the pGOI plasmid containing the ITR. The quantification was performed using the LightCycler system (Roche Diagnostic).
Protein interaction analyzer for detecting the concentration of recombinant adenovirus particles
The concentration of AAV843 particles (viral particles/mL, vp/mL) was detected by the Octet protein interactor (Fortebio). 20 μL of the test sample was added to 180 μL of PBST and mixed thoroughly; 180 μL of the above sample was added to 90 μL of PBST, mixed thoroughly, which was diluted a total of 15 times. 250 μL of standard substance (1.1 × 1010 vp/mL) was added to 250 μL of PBST, which was mixed thoroughly and diluted 7 times in a 1:1 ratio to obtain gradient dilution standard substance. The Octet protein interactor was performed according to the following steps: equilibrate at room temperature for 600 s, capture antibody for 180 s, buffer for 60 s, sample for 600 s, regeneration for 5 s, neutralization for 5 s, oscillation speed 1,000 rpm. The standard curve was fitted with the number of virus particles as the horizontal axis and the corresponding detection values as the vertical axis to obtain the fitting equation and R 2 value. The sample detection values were substituted into the fitting equation, then multiplied by the dilution factor, the concentration of virus particles in the sample was finally obtained. The relative full capsid ratio was generally calculated according to this formula: viral genome concentration (vg/mL) / viral particle concentration (vp/mL) × 100%.
Alkaline gel detection for FVIII AAV843 gene integrity
The viral genome was extracted from cell lysate using ViralDNA/RNA Purification Kit (Bioflux), as described in the user manual. The alkaline gel configuration method is as follows: add 0.8 g of agarose (Sangon Biotech) to 100 mL of ultrapure water, heat to dissolve, add NaOH to a final concentration of 50 mM, add EDTA to a final concentration of 1 mM, and cool to prepare alkaline gel. The preparation method of alkaline gel electrophoresis buffer is as follows: add 10 mL of 5M NaOH and 2 mL of 0.5M EDTA to 988 mL of purified water, mix well, and store at 2–8°C. Add 4 μL of loading buffer to 16 μL of sample and mix before loading, electrophorese with 100 V/220 mA for 5 min, and then add alkaline gel buffer, electrophorese with 75 V voltage for 1.5 h. Put the gel into 1×TAE and neutralize for 10 min. Put the gel into staining (100 mL 1×TAE with 20 μL GelRed nucleic acid gel stain). Incubate at room temperature for 1 h. Take photos using a gel imaging system (Protein Simple).
Detection of rAAV bioactivity
The bioactivity of rAAV expressing GFP was assayed by measuring the fluorescence intensity in infected HEK293 and Huh7 cells. The cell harvest solution was lysed using ultrasound, and the supernatant was collected after centrifugation at 12,000 g. Cells in a 96-well plate were infected with the supernatant at the same multiplicity of infection (MOI), and the green fluorescence intensity was measured 48 h later using a microplate reader (Molecular Devices) at an excitation wavelength of 475 nm and an emission wavelength of 525 nm. The bioactivity of the recombinant FIX rAAV was assayed using a coagulation time measurement method. 16 The viral harvest was subjected to ultrasonic lysis, followed by centrifugation at 12,000 g to collect the supernatant. An equal volume of the supernatant was then used to infect Huh7 cells at a consistent MOI. After a 48-h incubation period, the supernatant was collected to measure the coagulation time. This was compared with a standard reference coagulation time, which was set at 100%.
RESULTS
Simulating high-density perfusion culture of HEK293 cells in shake flasks
To ascertain the impact of varying CSPR on the proliferation of cells to higher densities, we set up different shake flask medium exchange volumes to simulate a range of CSPRs. The calculation formula employed was V = (ν1 + ν2)2 × CSPR × V1, where V represents the exchange volume, ν1 is the current cell density at the initiation of medium exchange when the cell density reaches 5 × 106 cells/mL, ν2 is the cell density expected after 24 h based on the normal cell doubling rate, typically on the first day (0–24 h) ν1 is 5 × 106 cells/mL, and 24 h after initiating perfusion, ν2 is 1 × 107 cells/mL. On the second day (24–48h), ν1 is 1 × 107 cells/mL and ν2 is 2 × 107 cells/mL, with V1 denoting the culture volume. The cell growth curves under different simulated CSPRs are depicted in Fig. 1A, and the daily doubling times are illustrated in Fig. 1B. The results indicated that from 0 h to 24 h after the commencement of centrifugal medium exchange, the cell doubling time progressively decreased with an increase in CSPR. Between 24 h and 48 h, the doubling time under simulated perfusion conditions approximated that of batch culture, suggesting that the cell state remained within the logarithmic growth phase. To ensure adequate nutrition post-transfection, a medium exchange volume of 0.05 μL/cell/day (40% of the culture volume on the first day and 80% on the second day) was selected as the simulated perfusion rate for subsequent optimization of transfection conditions in shake flasks.
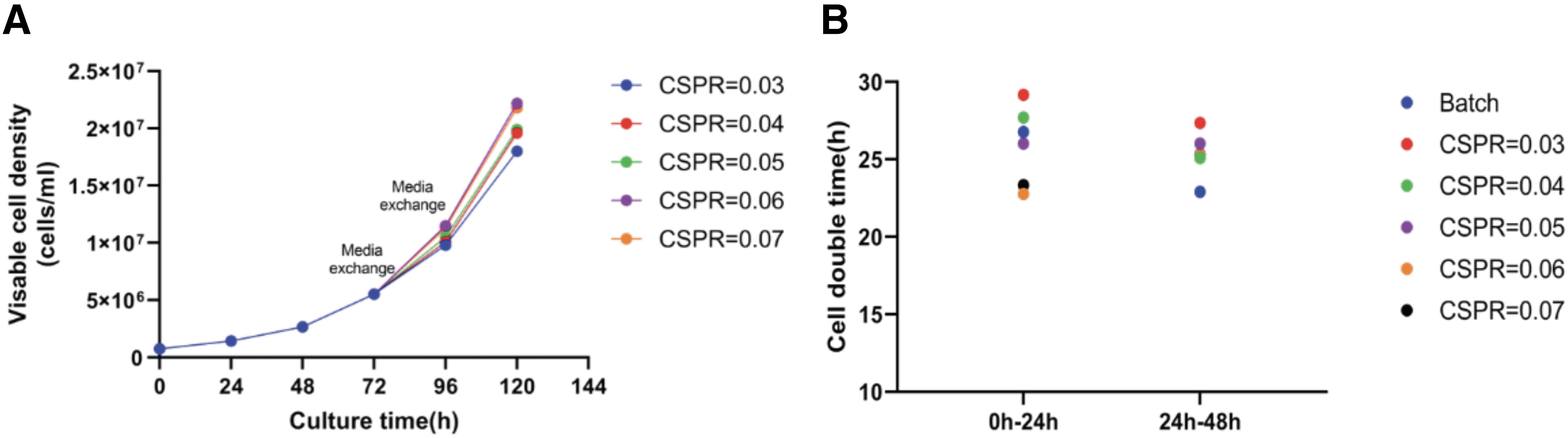
Growth curves and cell doubling times every 24 h of HEK293 cells under different cell-specific perfusion rates (CSPR) simulated by medium exchange in shake flasks.
High-density transfection for rAAV production in shaking flasks
To determine if a certain transfection efficiency could be achieved at high cell densities, initial transfection conditions were designed with cell densities of 1 × 107 cells/mL and 2 × 107 cells/mL. The amount of plasmid used was 0.5 µg corresponding to every 1 × 106 cells, with a DNA:PEI (w/w) ratio of 1:2. The mixture volume accounted for 10% of the total volume of the transfected cell solution and was incubated at room temperature for 4 min. Transfection was performed with pCMV-eGFP, followed by centrifugal medium exchange at 80% (CSPR = 0.05 μL/cell/day) to simulate perfusion. Transfection efficiency was assessed every 24 h using a Cellex cell counter, with results depicted in Supplementary Fig. S1. The findings indicated that at cell densities of 1 × 107 cells/mL and 2 × 107 cells/mL, transfection efficiencies could reach over 70%, similar to those under batch culture conditions. This suggests that transfection efficiency at high densities is not a bottleneck for virus packaging, although the optimal transfection conditions still warrant investigation. Clearly, at the same transfection efficiency, a cell density of 2 × 107 cells/mL would result in more cells being transfected to produce rAAV; hence, this density was used in subsequent studies.
To ascertain the optimal transfection conditions for packaging recombinant AAV at high densities, using eGFP AAV843 as the subject of study, a DoE was set for the amount of transfection plasmid, the DNA:PEI ratio, and the incubation time (Supplementary Figure S2). After transfection, centrifugal medium exchange at 80% (CSPR = 0.05 μL/cell/day) was used to simulate perfusion, and the cells were harvested 48 h post-transfection. The experimental results were fitted and analyzed using the least squares method. All single factor and binary interaction factors have p-values >0.05, and the response surface curve fitting graph is shown in Fig. 2A. Based on the response surface curve recommendations and actual transfection conditions, the optimal transfection conditions were determined to be a cell density of 2 × 107 cells/mL, a plasmid amount of 0.5 µg corresponding to every 1 × 106 cells, a DNA:PEI (w/w) ratio of 1:3, and a mixture volume accounting for 10% of the total volume of the transfected cell solution, with a 4-min incubation at room temperature. Under these conditions, the eGFP AAV843 virus yield was approximately 5 × 1011 vg/mL, compared with a typical yield of approximately 3 × 1011 vg/mL for fed-batch.

Optimization results of high-density transfection process simulated by medium exchange in shake flasks.
The impact of different simulated perfusion rates on the virus genome titer yield after transfection is shown in Supplementary Fig. S2. The results indicated that simulated perfusion rates between 0.05 and 0.1 μL/cell/day had a maximum virus production of about 6 × 1011 vg/mL, with a single cell production of only 3 × 104 vg/mL, which was lower than the fed-batch’s 6 × 104 vg/mL, indicating that the restrictive nutrition basic culture medium that affects virus packaging may not be able to provide it. Continuing to increase CSPR will consume a large amount of basic culture medium and have a lower cost-effectiveness. Therefore, the impact of high-concentration feed on virus yield was studied. Feeds that have been shown to increase virus yield in batch culture were tested, and the effects of feed volume and centrifugal medium exchange volume on virus yield are depicted in Fig. 2B. The results showed that in different rAAV packaging systems, not changing the medium (perfusion) after transfection but only adding feed could significantly increase virus yield, with eGFP AAV843 reaching a yield of 6.5 × 1011 vg/mL, over the medium exchange mode. These results may suggest that the feed contains nutritional factors not present in the basal medium that can enhance virus yield. The feed timing was also studied, with results shown in Fig. 2C. It was found that feeding 3 h post-transfection resulted in a virus yield of 1.1 × 1012 vg/mL, a 70% increase over feeding 24 h post-transfection, and 3.7 times the typical yield of fed-batch, with the single-cell yield under high-density conditions essentially reaching the level of fed-batch.
Subsequently, to confirm the robustness and practicality of the above process, the expression levels of other AAV serotypes packaging eGFP under the medium replacement simulated perfusion feed process were tested in shake flasks based on the aforementioned conditions, including AAV5, AAV8, and AAV9, with results shown in Fig. 3A. The results indicated that under high-density packaging conditions, the virus yields for other AAV serotypes reached 6.57 × 1011, 1.73 × 1012, and 1.07 × 1012 vg/mL, respectively, with single-cell yields also reaching levels comparable with those at low density, as shown in Fig. 3B. The same serotype (AAV843) packaging different genes, eGFP/recombinant FIX(rFIX)/rFVIII, was also tested, with results shown in Fig. 3C. The results indicated that the yields for the same serotype packaging different genotypes were significantly improved, with virus yields reaching 1.79 × 1012, 1.63 × 1012, and 1.17 × 1012 vg/mL, respectively, and single-cell yields also reaching levels comparable with those at low density, as shown in Fig. 3D. In addition, the in vitro activity of the recombinant viruses under the two culture modes was also studied. The results showed that under the fed-perfusion virus packaging process, the GFP activity of AAV843/AAV5/AAV8/AAV9 in Huh7 and HEK293 cells exhibited no significant difference in in vitro cellular activity compared with the fed-batch process; results are shown in Fig. 4A–D. The in vitro activity of the rFIX AAV843 produced under these conditions also showed no significant difference; results are shown in Fig. 4E. The full capsid ratio for the same serotype has also been studied, and the results show that there is no significant difference in full capsid ratio between the two production methods for AAV843 serotype rAAV cell lysates of different gene sizes (Supplementary Fig. S3).

Comparison of total and per-cell yields of different types of rAAV produced by the fed-batch and fed-perfusion processes in shake flasks.

Comparison of the in vitro activity of different AAV serotypes produced by the fed-batch and fed-perfusion processes in shake flasks.
The comparison between the Fed-batch culture process in shake flasks and the high-density transfection and feeding process using simulated perfusion is shown in Fig. 5. The cell seeding density at the beginning and the cell proliferation process after 1–2 days of the two cultivation processes is the same. Fed-batch culture is performed transfection on the third day, when the cell density reaches 3-5e6 cells/ml. The fed-perfusion process used the perfusion process to further culture the cells to a higher density of 1.8-2.2e6 cell/ml before transfection. In the subsequent two processes, the feed process was used. But the volume of filling feed is different.
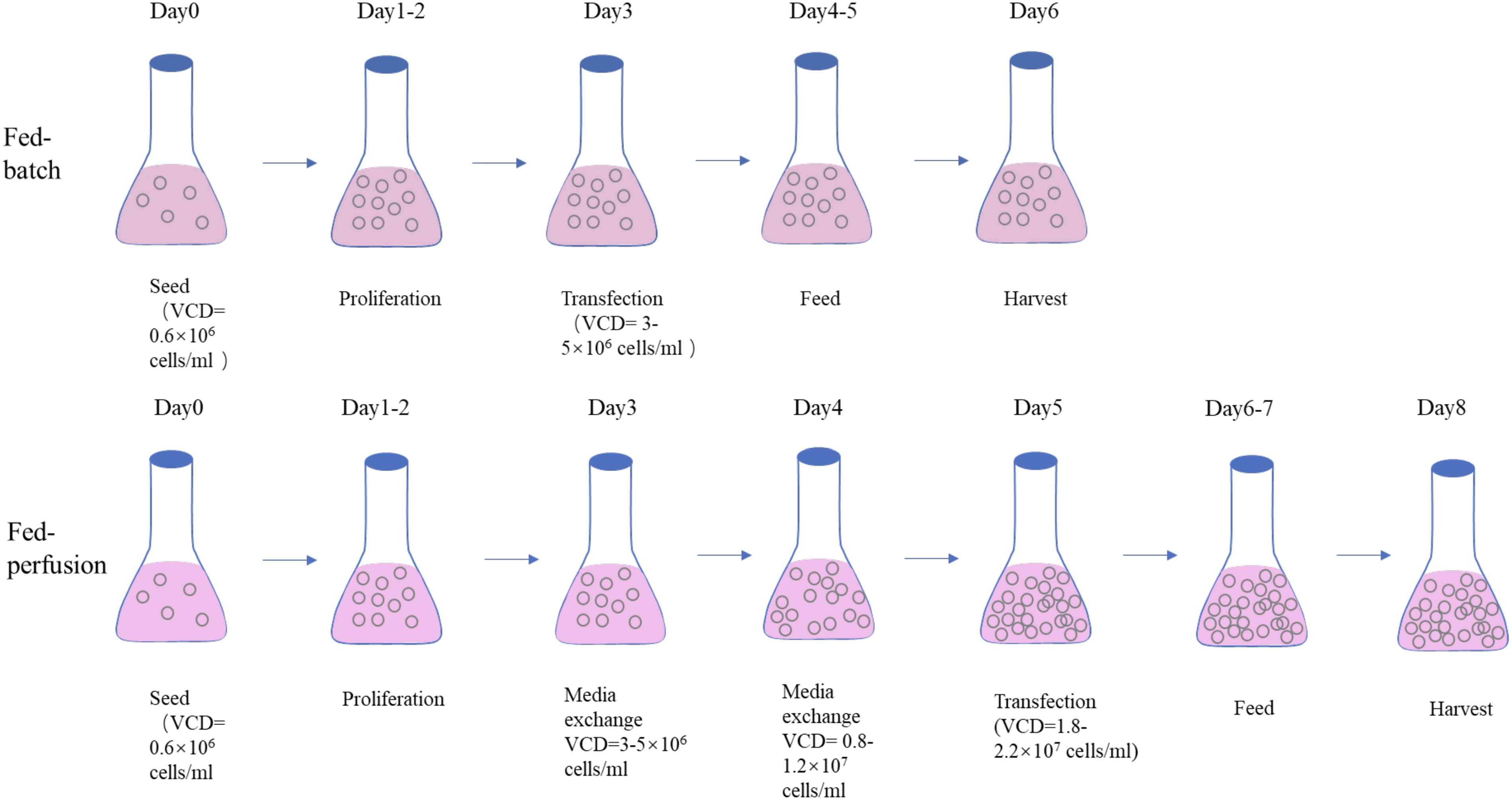
Comparison of the low-density process by fed-batch and the high-density process simulated by medium exchange in shake flasks for HEK293 cells. This high-density production process is named fed-perfusion.
Perfusion high-density culture of HEK293 cells in a 3 L bioreactor
To achieve a scalable high-density perfusion transfection process for the production of rAAV, a Biosep acoustic cell separation device was utilized to separate cells from the culture medium for perfusion culture. By monitoring the cell density in the effluent, it was observed that the Biosep maintained a cell separation efficiency of over 95% for cell densities below 2 × 107 cells/mL (data not shown), ensuring the feasibility of the perfusion culture approach. The perfusion rate is a critical factor influencing cell growth; a lower perfusion rate that supports normal cell growth represents an optimal use of the culture medium. However, a lower growth rate may impact virus production. The curves of different perfusion rate viable cell densities and viabilities over time in the 3 L bioreactor are depicted in Fig. 6A, with cell doubling times presented in Fig. 6B. The results showed that with an initial inoculation at 0.8 × 106 cells/mL, the doubling time was longer, approximately 30 h, during the first day of perfusion (Day 4) when the CSPR was 0.04 μL/cell/day, and this doubling time gradually decreased with an increase in CSPR. All CSPRs exhibited no significant difference in doubling time on the second day following the initiation of perfusion (Day 5), with all being within the range of 26–28 h. By around 124 h, the maximum cell density could reach over 1.8 × 107 cells/mL regardless of the CSPR used. It is noteworthy that, potentially due to the properties of the culture medium, when perfusion was initiated after the cell density exceeded 5.5 × 106 cells/mL, the cell doubling time increased substantially with any of the aforementioned CSPRs. It took over 96 h for the cell density to increase to 2 × 107 cells/mL, resulting in a lower virus yield post-transfection (data not shown).
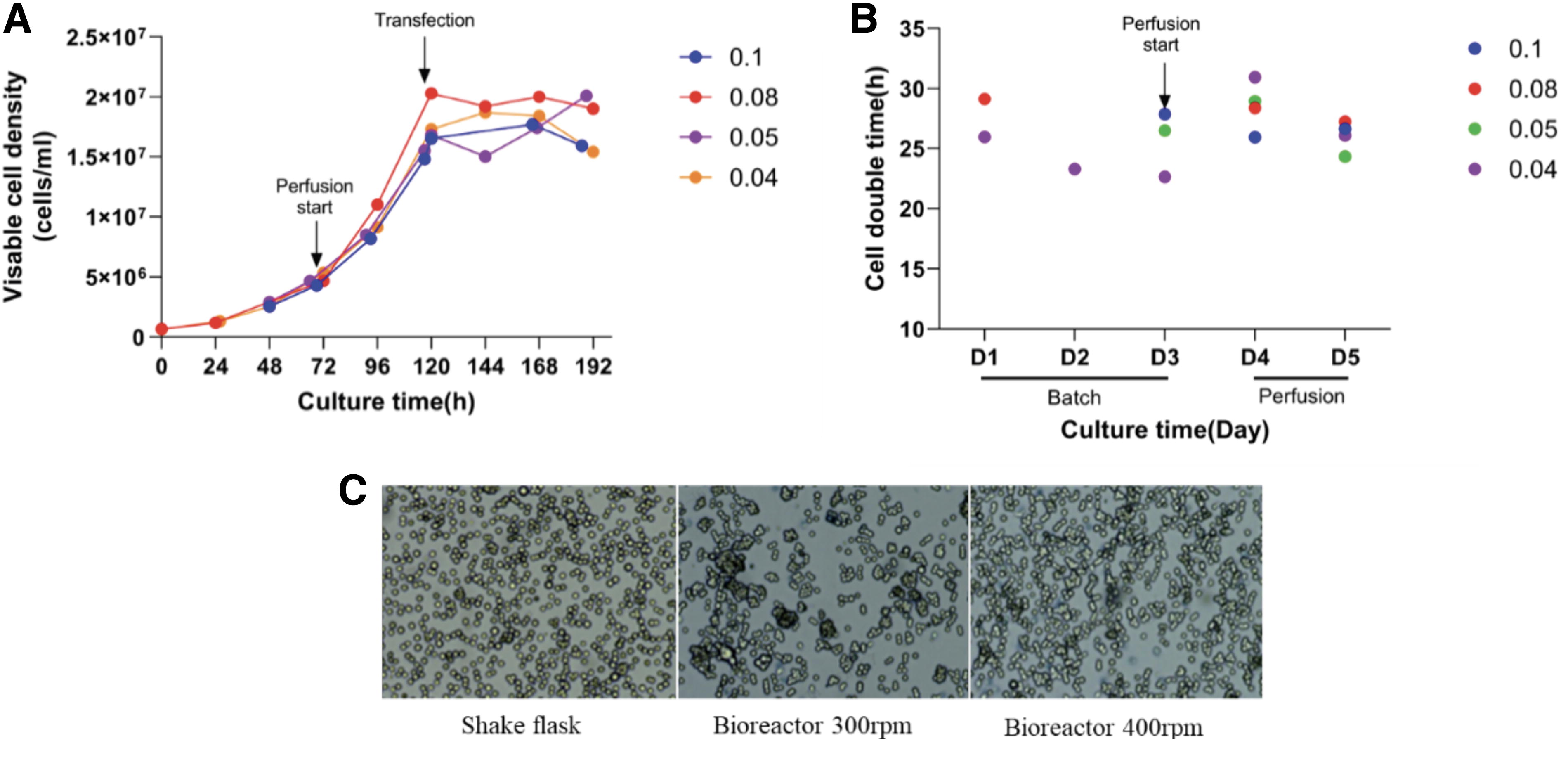
Growth curves, doubling times, in a 3 L bioreactor with different CSPRs and cell status of HEK293 cells with different rpm using Biosep for perfusion culture.
High-density transfection for rAAV production in a 3 L bioreactor
Under fed-batch conditions (Viable cell density, VCD ≈ 5 × 106 cells/mL), the yield in the 3 L bioreactor was essentially equivalent to that in shake flasks. Upon reaching a high cell density (VCD ≈ 2 × 107 cells/mL) through normal cell doubling via perfusion culture, the cells were transfected with the optimal transfection conditions previously established in shake flasks for the packaging and expression of rFVIII, with the capsid being AAV843. The initial conditions set were a rotation speed of 300 rpm and a CSPR of 0.8 μL/cell/day, employing an optimized feed-perfusion process (Condition 1). The yield, however, was only 4.6 × 1011 vg/mL, which did not meet the high-density shake flask packaging yield of over 1 × 1012 vg/mL. The single-cell yield was merely 0.22 × 105 vg/cell, indicating the presence of limiting factors in the transfection and virus packaging stages of the process.
To identify the limiting factors at the transfection stage, eGFP was transfected under the Condition 1 to assess transfection efficiency. The transfection efficiency after 48 h was only 43%, suggesting the existence of limiting factors under these conditions. A comparison of cell morphology during transfection between the bioreactor and shake flasks revealed more cell clumping in the bioreactor, whereas less clumping was observed in the shake flasks. This could be attributed to the increased propensity for cell aggregation within the bioreactor, exacerbated by the extended culture time at high density, leading to reduced transfection efficiency. The addition of an anti-clumping agent is an effective method to counteract cell clumping, but the incorporation of such agents has been found to significantly decrease transfection efficiency upon research.
An alternative approach to reducing cell clumping is to enhance the agitation intensity within the bioreactor by increasing the rotation speed. When the rotation speed of the reactor was increased, the cell agglomeration during transfection was shown in Fig. 6C. The degree of cell agglomeration was low in the simulated perfusion culture in the shaking flask. In the perfusion culture of the bioreactor, there were more agglomerations under the condition of 300 rpm, and the cell agglomeration during transfection was significantly decreased after the speed was increased to 400 rpm (Condition 2). With the increased rotation speed, the 48-h transfection efficiency in the bioreactor rose to 76%, and the virus yield increased to approximately 6 × 1011 vg/mL, representing a 30% improvement over the lower rotation speed, as detailed in Table 1. This indicates that cell clumping at high densities can significantly impact transfection efficiency, thereby limiting virus yield. However, the virus yield in the bioreactor still did not reach the desired yield, suggesting that other limiting factors are at play.
Production of rAAV under fed-perfusion process on a 3 L stirred bioreactor
Comparison of unit volume yield and -ell yield of rFXIII AAV843 under different paddle speeds and CSPR and the same process under shake flask and bioreactor using the fed-batch process.
CSPR, cell-specific perfusion rates; rAAV, recombinant adeno-associated virus.
In shake flasks, daily medium exchange simulated perfusion translates to a CSPR of approximately 0.05 μL/cell/day rather than 0.08 μL/cell/day in bioreactor, which may suggest that varying CSPRs can affect transfection efficiency. Under Condition 2, when the CSPR was reduced to 0.05 μL/cell/day and increased to 0.1 μL/cell/day, the results are presented in Table 1. The findings indicate that both lowering and raising the CSPR can enhance virus yield, with a single-cell virus yield of 0.56 × 105 vg/cell at CSPR = 0.1 μL/cell/day and 0.46 × 105 vg/cell at CSPR = 0.05 μL/cell/day. This may imply that different CSPRs have an impact on cell status and transfection efficiency. A lower CSPR of 0.04 μL/cell/day was also tested, with single-cell virus yields comparable with those at CSPR = 0.05 μL/cell/day and CSPR = 0.1 μL/cell/day. Ultimately, after optimization of rotation speed and CSPR, the single-cell yield in the bioreactor essentially reached the level of the shake flasks (0.55 ± 0.06 × 105 vg/cell) and the bioreactors using fed-batch process (0.60 ± 0.04 × 105 vg/cell), as shown in Table 1. The partial genome packaging of FVIII AAV843 under two production processes was verified through alkaline gel experiments to determine the effect of partial genome packaging on virus titer. The results are shown in Supplementary Fig. S4, which shows that the partial genome under the fed-batch process is basically the same as that under the fed-perfusion process. Due to the high yield under the fed-perfusion process, the FVIII genome band can be seen at 5.0 kb.
DISCUSSION
This study has demonstrated that the pre-mixing method of plasmid and PEI can efficiently transfect cells at high densities, with transfection efficiencies reaching over 80% at 2 × 107 cells/mL, laying the foundation for subsequent high-density packaging of rAAV using a three-plasmid system. The DOE for the three-plasmid transfection conditions has identified the optimal conditions for this process. The ratios of the three plasmids were not investigated, as it was considered that the transfection efficiency, which is the primary factor affecting virus yield at high density, is mainly influenced by the amount of plasmid used, the DNA:PEI ratio, the mixing volume, and the incubation time. Therefore, the optimal conditions from low-density transfection were used for the three-plasmid ratio.
The transition from shake flask to bioreactor represents a significant variable in the process that affects the subsequent methods for high-density transfection and packaging of rAAV, determining whether high-density processes can be scaled up for broader application. Currently, perfusion culture technology has become relatively mature, and this study has also verified that HEK293 cells can be cultured to higher densities through perfusion. Additionally, some important factors affecting virus packaging in bioreactors have been identified, such as cell clumping and perfusion rate. Cell clumping can be addressed by increasing the agitation speed, which also improves the oxygen transfer rate (KLa) in the bioreactor, thereby reducing the power input for gases and saving resources. However, the improvement in cell clumping at higher scales due to increased speed may be limited, and further reduction in cell clumping can be achieved by screening for cell lines with a lower tendency to clump or by improving the composition of the culture medium. The impact of perfusion rate on virus yield may be due to specific perfusion rates affecting the composition of the culture medium during transfection, thereby influencing transfection efficiency and cell status, leading to lower virus yields. Further research can be conducted in this area. However, a low CSPR (0.05 μL/cell/day) enhances the cost-effectiveness of high-density transfection, with the additional 2 days only requiring 1.125 times more culture volume but yielding an additional 3–4 times more virus, improving the utilization rate of the culture medium.
The Biosep based on acoustic separation and ATF or TFDF are currently widely used perfusion systems. Both ATF and TFDF have been proven to culture HEK293 cells to high densities and successfully perform three-plasmid transfections, indicating that the choice of perfusion equipment during the cell proliferation phase may not be a limiting factor. Perfusion based on ATF or TFDF requires the use of high-value consumables, while perfusion culture based on Biosep only requires stainless steel fixed devices that can be cleaned and reused multiple times, potentially giving Biosep an advantage in terms of cost for large-scale production. A disadvantage of Biosep at high densities may be the reduced separation efficiency at densities exceeding 2 × 107 cells/mL, but the operational space for transfection at higher densities using the pre-mix method is also more limited, such as shorter mixing times while maintaining a constant mixing volume, as a higher mixing volume (>15% of the cell culture volume) may dilute the cell fluid, reduce virus yield, and increase the time for pumping the mixture into the culture, which is not conducive to large-scale cultivation.
One challenge in scaling up high-density transfection is the short incubation time after mixing plasmid and PEI, as seen in this study where the incubation time for high-density transfection was only 4 min, posing a challenge for large-scale production. This could be addressed by batch transfection or using variable mixing and addition equipment. Another challenge is the high host cell density after cell lysis at high densities, as virus vectors with poor secretion capabilities require cell lysis to release the virus. This could potentially be resolved by diluting the cell lysate or increasing the chromatography volume, but further research is needed in this regard.
CONCLUSIONS
In this study, we ameliorated the rAAV productivity in suspension-adapted HEK293 cells by optimizing culture, perfusion, and transfection conditions, particularly at high density of 2 × 107 cells/mL. With preferable transfection parameters and feeding regimen, we managed to reach a volumetric yield of 1–2 × 1012 vg/mL in flasks without compromising the unit yield per cell and bioactivity of the viral products. Subsequently, this DOE methodology was recapitulated and further adjusted to a 3 L bioreactor, pinpointing key factors that herein might influence the viral yield comprising the initial cell density, cell aggregation, and perfusion rate. Significantly, we actualized a 3-fold increase in the yield of an AAV vector encapsulating oversized rFVIII sequence of 5.1 kb, which exerts great challenge on manufacture. The yield of this vector was raised to over 9 × 1011 vg/mL, which is a substantial enhancement over the conventional fed-batch process. These findings not only pave the way for a scalable high-density transfection technique for rAAV production but also provide a solid foundation for future large-scale applications in the field.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge Shou-Bo Hu for his support in Biosep and bioreaction and Rui-Xia Wang, Chun-Tao Pang, and Dao-Shu Tan for their technical support.
AUTHORS’ CONTRIBUTIONS
Z.D.: Conceptualization, methodology, formal analysis, part of investigation, data curation, writing—original draft preparation, and visualization. Y.-L.L.: Resources, part of investigation, validation, and data curation. X.-T.W. and L.-H.Y.: Resources and part of investigation. K.Z.: Writing—reviewing and editing. Z.-M.D.: Supervision and validation. X.X.: Supervision and funding acquisition.
DATA AVAILABILITY STATEMENT
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the National Key Research and Development Program of China (No. 2021YFC2700803).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.