
Abstract
The low-density lipoprotein receptor (LDLR) plays a crucial role in cholesterol regulation and lipoprotein transport. Variations in the LDLR gene can cause familial hypercholesterolemia (FH), with homozygous familial hypercholesterolemia (HoFH) being the most severe form. HoFH is marked by elevated low-density lipoprotein cholesterol (LDL-C) levels and early onset of cardiovascular disease, often with a poor prognosis. Current treatment options for HoFH are limited by insufficient effectiveness and restricted availability. Gene therapy, which involves the delivery of functional LDLR genes, offers a promising and innovative approach that could significantly improve outcomes for patients with HoFH. In this study, the adeno-associated virus serotype 8 (AAV8) vector was used to deliver the LDLR gene specifically to hepatocytes. The vector was designed using the pAAV-TBG plasmid, incorporating a hepatocyte-specific thyroid hormone-binding globulin (TBG) promoter. Viral packaging was performed in HEK 293T cells, followed by virus collection, purification, and titration. Mice, including C57BL/6J, Ldlr-KO, and homozygous Ldlr p.W483X mice, were injected with low, medium, or high doses of the virus via the tail vein. The efficacy and safety of the AAV8-LDLR gene therapy were assessed through Western blot analysis, lipid profiling, and liver pathology. AAV8-mediated LDLR delivery effectively improved lipid levels in both Ldlr-KO and homozygous Ldlr p.W483X mice. LDL-C levels showed a sustained reduction over the 2-month observation period. Western blot analysis confirmed the expression of LDLR protein in the liver, while lipid profiling demonstrated significant reductions in total cholesterol, triglycerides, LDL-C, and high-density lipoprotein cholesterol levels. Liver histopathology revealed no significant differences in non-alcoholic fatty liver disease scores between groups, indicating a favorable safety profile, particularly at low and medium doses. AAV8-LDLR gene therapy shows considerable promise as an effective treatment for HoFH. Our results indicate that this therapy significantly reduces lipid levels while maintaining a favorable safety profile.
INTRODUCTION
The low-density lipoprotein receptor (LDLR) is a key cell surface glycoprotein responsible for regulating the transport of lipoproteins. 1 It facilitates cellular uptake by binding to apolipoprotein E (apoE) and apolipoprotein B (apoB) present in low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL), maintaining cholesterol homeostasis. 2 Variations in the LDLR gene can reduce or eliminate LDLR expression on the cell surface, impairing cholesterol metabolism and leading to elevated blood LDL cholesterol (LDL-C) levels. This dysregulation can result in lipid metabolism disorders, including familial hypercholesterolemia (FH), which occurs in two forms: heterozygous FH (HeFH) and the more severe homozygous FH (HoFH). 3
HoFH is a rare and serious genetic disorder characterized by extremely high LDL-C levels and a high risk of premature cardiovascular disease, often caused by biallelic LDLR inactivation. 4,5 Despite existing treatments, effective and accessible options for HoFH remain limited. Current management includes lifestyle changes, cholesterol-lowering medications, lipoprotein apheresis, and liver transplantation. 6 However, lifestyle interventions alone are typically insufficient, requiring patients to use cholesterol-lowering drugs. Guidelines recommend high-intensity statins with ezetimibe as the first line of therapy, with the addition of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors such as evolocumab or alirocumab if necessary. However, these therapies often have limited efficacy, reducing LDL-C levels by only about 30%, leaving most patients with elevated levels. 7,8
Non-LDLR-dependent treatments such as lomitapide (a microsomal triglyceride transfer protein inhibitor) and evinacumab, an angiopoietin-like protein 3 (ANGPTL3) inhibitor can lower LDL-C by 40–60%, but both have drawbacks. Lomitapide can cause side effects, including liver steatosis, elevated transaminases, and gastrointestinal issues. 9 Evinacumab, approved only for patients aged 12 and older, is expensive and requires monthly intravenous administration. 10 Other treatments, such as lipoprotein apheresis, offer only short-term reductions in LDL-C and are associated with a significant impact on quality of life and financial burden. 11 Liver transplantation, though effective, is limited by high costs, donor shortages, surgical risks, and the need for lifelong immunosuppressive therapy. 12
Recent advances in adeno-associated virus (AAV)-based gene therapy offer new hope for the treatment of HoFH. 13 By using AAV vectors to deliver a functional LDLR gene to the liver, sustained LDLR expression can be achieved, resulting in reduced serum LDL-C levels and prevention of atherosclerotic cardiovascular disease. In 1994, a 29-year-old female underwent the first ex vivo gene therapy attempt for HoFH in humans. 14 Shortly thereafter, in 1995, Grossman et al. 15 conducted a pilot study using the same ex vivo approach, transplanting a limited number of retrovirus-transduced hepatocytes that achieved continuous gene expression for at least 4 months, providing a novel perspective on liver-targeted gene therapy. However, this method showed limited improvement in LDL-C levels, and its efficacy declined over time. In 2004, Lebherz et al. 16 compared recombinant AAV vectors expressing human LDLR in Ldlr-knockout (KO) mice and found that adeno-associated virus serotype 8 (AAV8) outperformed AAV2 and AAV7 in reducing hyperlipidemia and atherosclerosis (AS), with longer-lasting effects. In 2016, the first phase 1/2 clinical trial of AAV-mediated human LDLR gene transfer was conducted in nine HoFH patients (NCT02651675); however, some patients experienced elevated liver transaminase levels after treatment. In contrast, a 2018 study investigating AAV8 gene therapy for Crigler–Najjar syndrome in rhesus macaques supported the clinical translation of AAV8, demonstrating no severe hepatotoxicity, innate immune activation, or systemic inflammation. 17 In addition, AAV8-based gene delivery therapies have demonstrated significant therapeutic potential in clinical trials for glycogen storage disease type Ia. 18 These successes suggest continued potential for AAV8-mediated therapy for HoFH, although further optimization of dose and vector design remains necessary.
In this study, we synthesized the LDLR gene and incorporated it into an AAV8 vector with liver-selective specificity. We evaluated this gene therapy approach in LDLR-KO mice and a mouse model carrying the patient-derived p.W483X homozygous point variation to explore its potential as a treatment for HoFH.
METHODS
Vector construction
The pAAV-TBG vector was used to construct the AAV vectors, utilizing the hepatocyte-specific thyroid hormone-binding globulin (TBG) promoter to ensure targeted gene expression. The target gene was preceded by the Kozak sequence GCCACC and was expressed without any additional tags or fluorescent markers.
Virus packaging
HEK 293T cells were prepared, and the growth medium was replaced with serum-free Dulbecco’s dodified eagle medium (DMEM) containing 1% 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) and 1% penicillin/streptomycin 1–2 h before transfection. A transfection mixture was prepared with a ratio of 15:2:2:1 for the transfection reagent, packaging plasmid, vector plasmid, and helper plasmid, respectively. This mixture was incubated at room temperature for 30 min before being added to the cells. The cells were gently mixed by shaking and then incubated at 37°C with 5% CO2 for 72 h to allow for virus packaging.
Virus collection
After transfection, the cells were lysed, and the lysate was centrifuged to separate the cell pellet from the supernatant. The supernatant was transferred to a new tube and mixed with polyethylene glycol (PEG) 8000 precipitant solution (2.33 g NaCl and 8.5 g PEG 8000 per 100 mL) for overnight precipitation. The next day, the mixture was centrifuged at 3,500 g for 30 min at 4°C. The supernatant was discarded, and the pellet was resuspended in phosphate-buffered saline (PBS) containing 0.001% Pluronic F-68 (PF68). The cell pellet was also resuspended in the same buffer, subjected to a single freeze–thaw cycle, and supplemented with 5M NaCl. The mixture was vortexed to ensure thorough mixing. The supernatant and resuspended pellet were combined and sonicated to achieve a uniform, non-viscous consistency. Sonication conditions varied based on the viscosity, with three to four pulses for 30% amplitude (AMPL) and a single pulse for 20% AMPL. After sonication, the mixture was centrifuged at 3,500 g for 30 min, and the supernatant was collected for further use.
Virus purification
Iodixanol gradients with decreasing concentrations were prepared in an ultracentrifuge tube, starting with the highest concentration at the bottom and carefully layering the lower concentrations above to avoid mixing. The virus solution was then carefully layered on top of the gradient. The virus was purified through ultracentrifugation.
Virus titer and specificity assay
To determine the virus titer, the viral capsid was broken down using a mixture of 5 μL of virus solution, 1 μL of proteinase K (5 μg/μL), and 4 μL of ultrapure water. This mixture was incubated at 37°C for 30 min and then heated at 95°C for 5 min to inactivate the enzyme. The solution was centrifuged at 1350 g for 2 min, and the supernatant was collected.
For quantification, PCR was performed using a 20 μL reaction mixture containing 10 μL of 2× SYBR Green Mix, 7.2 μL of ultrapure water, 0.8 μL of forward/reverse (F/R) primer mixture, and 2 μL of virus sample or standard. The PCR conditions were as follows: 95°C for 5 min, followed by 40 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s. The virus titer was calculated based on the standard dilution curves.
To assess specificity, a separate 20 μL PCR reaction was prepared with 1.5 μL of quantitative PCR product, 10 μL of 2× Mix, 4 μL of F/R primer mixture, and 4.5 μL of ultrapure water. The PCR protocol included the following: 95°C for 4 min, followed by cycles of 95°C for 30 s, 64°C for 45 s, and 72°C for 1 min (1 cycle); 95°C for 30 s, 62°C for 45 s, and 72°C for 1 min (1 cycle); and 95°C for 30 s, 60°C for 45 s, and 72°C for 1 min (35 cycles), with a final extension at 72°C for 10 min. The PCR products were analyzed by gel electrophoresis to compare band sizes and confirm specificity.
Animal models
Ldlr p.W483X knock-in mice were generated using clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated 9 (Cas9) systems. Fertilized eggs were co-injected with the guide RNA (gRNA) targeting the mouse Ldlr gene, Cas9 protein, and donor oligonucleotide containing the TGG to TAG mutation. Founders (F0) were validated by PCR and sequencing and then bred with wild-type C57BL/6J mice to establish germline-transmitted F1 progeny.
Gene therapy studies
Study 1: AAV8-LDLR in Ldlr-KO mice
Age-matched male Ldlr-KO and C57BL/6J mice (6–8 weeks) were acclimatized for 3 days followed by 4–5 h fasting for baseline lipid profiling (total cholesterol [TC], triglycerides [TG], LDL-C, and high-density lipoprotein cholesterol [HDL-C]). After stratified randomization by LDL-C levels, mice received intravenous AAV8-LDLR at three doses: 5 × 1011 (low), 1 × 1012 (medium), or 2 × 1012 vector genomes (vg)/mouse (high). Cohorts were maintained for 8 weeks on normal chow (CD) or 60% high-fat diet (HFD) with weekly weight and LDL-C monitoring. Terminal analyses included lipid profiling, liver hematoxylin and eosin (H&E) staining, and Western blot.
Study 2: Comparative phenotype analysis
Age-matched male mice (6–8 weeks), including Ldlr p.W483X homozygotes, Ldlr-KO, and C57BL/6J, were subjected to a 12-week dietary intervention (CD vs. 60% HFD) post-acclimatization. Longitudinal monitoring of weight and lipid parameters was performed weekly. Terminal analyses included lipid profiling, liver H&E staining, and Western blot.
Study 3: AAV8 efficacy in Ldlr p.W483X mice
Age-matched male Ldlr p.W483X and C57BL/6J mice (6–8 weeks) underwent 4 weeks of HFD preconditioning followed by low-dose AAV8-LDLR administration (5 × 1011 vg/mouse). Continuous HFD feeding for 8 weeks was maintained with weekly phenotyping. Terminal analyses included lipid profiling, liver H&E staining, and Western blot.
All procedures were approved by the Institutional Animal Care and Use Committee (Protocols: GPTAP20230322-3, GPTAP20231106-1, and GPTAP20240304-1).
Western blot analysis
To extract total proteins, the appropriate volume of prechilled radioimmunoprecipitation assay buffer (RIPA) lysis buffer, supplemented with protease inhibitors, was added to the samples. The mixture was homogenized, lysed on ice, and then centrifuged to obtain the supernatant, which was stored at −80°C. Protein concentration was determined using the bicinchoninic acid assay (BCA) protein assay kit.
For gel electrophoresis, 6% separating gel and 5% stacking gel were prepared. Each well was loaded with 25 µg of total protein. The proteins were separated using a constant voltage of 85 V until the completion of the run. The membrane was then transferred at constant pressure for 100 min. Following transfer, the membrane was incubated overnight at 4°C with a primary antibody specific for LDLR (polyclonal, diluted 1:2,000). After three washes with Tris-buffered saline with Tween-20 (TBST) (5 min each), the membrane was incubated with a secondary antibody (Goat anti-Rabbit IgG, horseradish peroxidase [HRP]-conjugated, diluted 1:10,000) at room temperature for 1 h. The membrane was then washed three times with TBST (5 min each) before developing the image using enhanced chemiluminescence. Details of the antibodies used are listed in Supplementary Table S4.
H&E staining
Tissues were fixed overnight in 4% paraformaldehyde and then processed through an automatic dehydrator to complete dehydration. Embedding was performed using an embedding machine, and sections were cut to a thickness of 3 µm using a paraffin microtome. The slides were then dried in an oven at 37°C.
The paraffin sections were deparaffinized using an environmentally friendly dewaxing solution (I and II), each for 10 min. The sections were then sequentially immersed in 100%, 95%, 85%, 70%, and 50% ethanol, with 3 min at each concentration, followed by a 5-min rinse in pure water.
Next, the sections were stained with hematoxylin for 3 min, washed three times with tap water, differentiated in 1% hydrochloric acid ethanol for 3–5 s, and washed again with tap water. The sections were then treated with 1% ammonia for 20–30 s to return to blue, followed by additional washes with tap water.
Dehydration was carried out sequentially in 50%, 70%, 85%, and 95% ethanol, each for 30 s, and then stained with eosin for 7 min. Further dehydration was done using 95% and 100% ethanol (I and II), each for 5 min. The sections were cleared using environmentally friendly dewaxing solution (I and II) for 5 min each.
Finally, excess dewaxing solution was wiped off, and an appropriate amount of neutral gum was applied before sealing with a coverslip.
Enzyme-linked immunosorbent assay
Serum concentrations of oxidized low-density lipoprotein (oxLDL) and C-reactive protein (CRP) were quantified using commercial enzyme-linked immunosorbent assay (ELISA) kits specific for mouse targets (oxLDL: CSB-E07933m, CRP: CSB-E07923m; CUSABIO, Wuhan, China) in strict accordance with the manufacturer’s protocols. The experimental procedure was conducted as follows: Standards and serum samples were loaded into antibody-precoated microplates and incubated at 37°C for 2 h. Following incubation, the liquid was decanted, and wells were washed three times. Subsequently, biotin-conjugated detection antibody was added to each well, followed by a 1-h incubation at 37°C. After repeating the washing procedure, HRP-conjugated avidin was introduced into all wells and incubated for 1 h at 37°C. Following five additional washing cycles, tetramethylbenzidine substrate solution was added per well and developed in the dark at 37°C for exactly 30 min. The enzymatic reaction was terminated by adding the stop solution. Optical density values were immediately measured at 450 nm.
Statistical analysis
Data were analyzed with GraphPad Prism (version 10.2.3) or SPSS (26.0) and presented as mean ± standard error of the mean. For statistical comparisons, we initially evaluated data for normality and equality of variances. Statistical analyses were performed using one-way analysis of variance (ANOVA) for multiple-group comparisons. If equal variances were not assumed, Welch’s ANOVA was applied. For non-normally distributed data, a non-parametric Kruskal–Wallis test was used for multiple-group comparisons. Statistical significance was set at p < 0.05.
RESULTS
Effectiveness of AAV8-LDLR gene therapy in Ldlr-KO mice
To evaluate the potential of AAV8-LDLR gene therapy for homozygous familial hypercholesterolemia (HoFH), we established a drug efficacy evaluation system using Ldlr-KO and C57BL/6J mice (Fig. 1A). The TBG promoter allows for targeted expression of the LDLR vector specifically in hepatocytes, thereby compensating for the deficient LDLR levels in these cells. AAV8-LDLR was administered intravenously to Ldlr-KO mice, and we included an empty vector control group (Group G3), as well as low (5 × 1011 vg/mouse, Group G4), medium (1 × 1012 vg/mouse, Group G5), and high (2 × 1012 vg/mouse, Group G6) dose treatment groups to assess the therapy’s efficacy and identify the optimal dose.
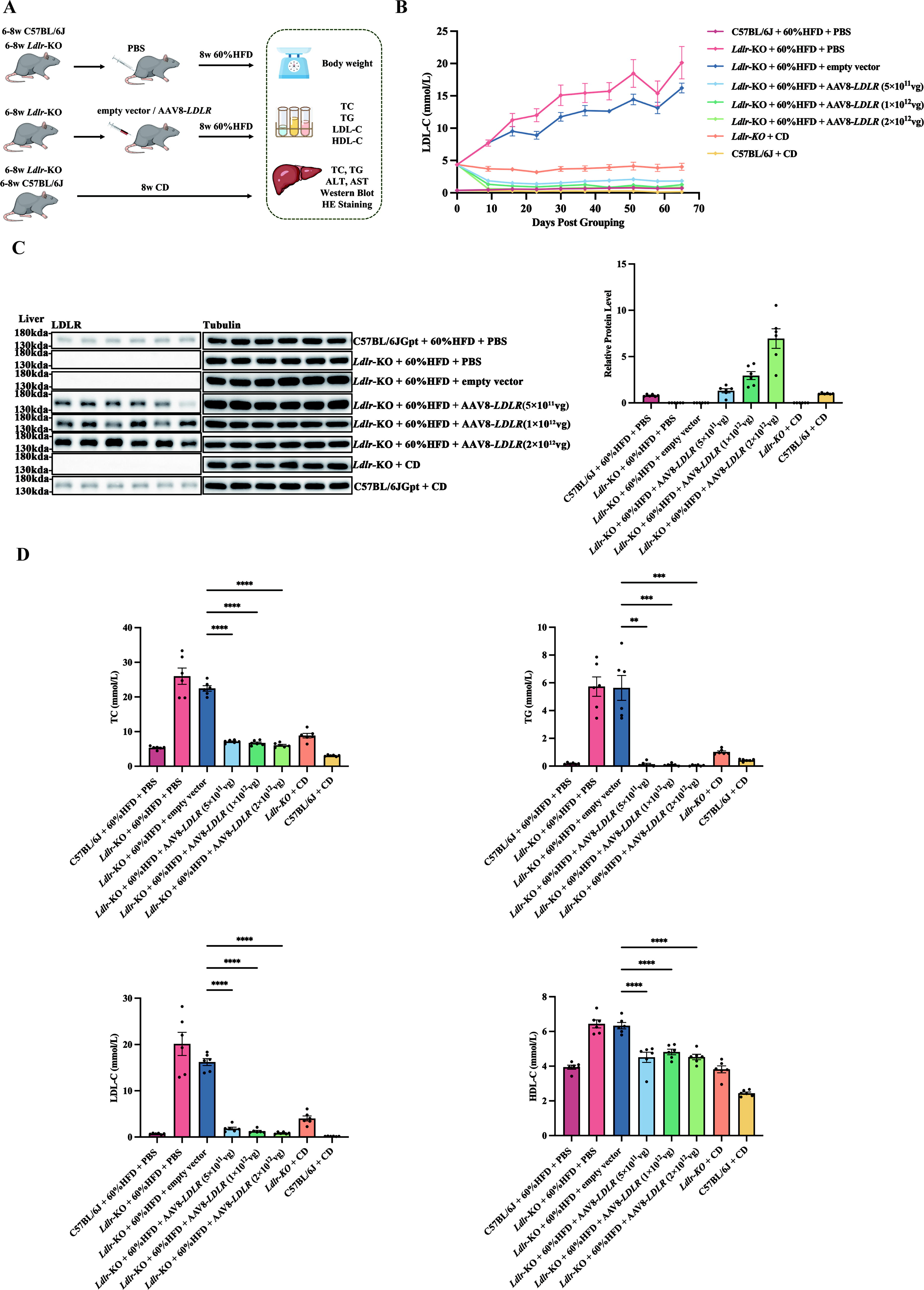
AAV8-LDLR gene therapy reduces hyperlipidemia in Ldlr-KO mice.
The serum LDL-C levels demonstrated that AAV8-LDLR effectively reduced LDL-C and maintained low levels throughout the 2-month observation period. The therapeutic effect improved with increasing concentrations of the vector (Fig. 1B). Western blot analysis confirmed that the LDLR protein expression in the liver was restored in a dose-dependent manner, compensating for the loss of LDLR expression in Ldlr-KO mice (Fig. 1C). Additionally, at the study’s endpoint, serum lipid levels, including TC, TG, LDL-C, and HDL-C, were significantly reduced in the Ldlr-KO mice treated with AAV8-LDLR (Fig. 1D). These results indicate that AAV8-LDLR gene therapy is a promising approach for managing refractory hyperlipidemia such as HoFH.
Safety profile of AAV8-LDLR gene therapy
To assess the safety of AAV8-LDLR gene therapy, liver tissues from treated mice were analyzed using H&E staining (Fig. 2A). We evaluated non-alcoholic fatty liver disease (NAFLD) based on hepatocyte steatosis, lobular inflammation, and ballooning degeneration (Supplementary Table S5). The results indicated a reduction in the NAFLD scores and NAS for the low-dose and medium-dose groups compared to the empty vector control group. Although the high-dose group had a higher NAFLD score and NAS, statistical analysis revealed no significant differences between the groups Fig. 2B, C.
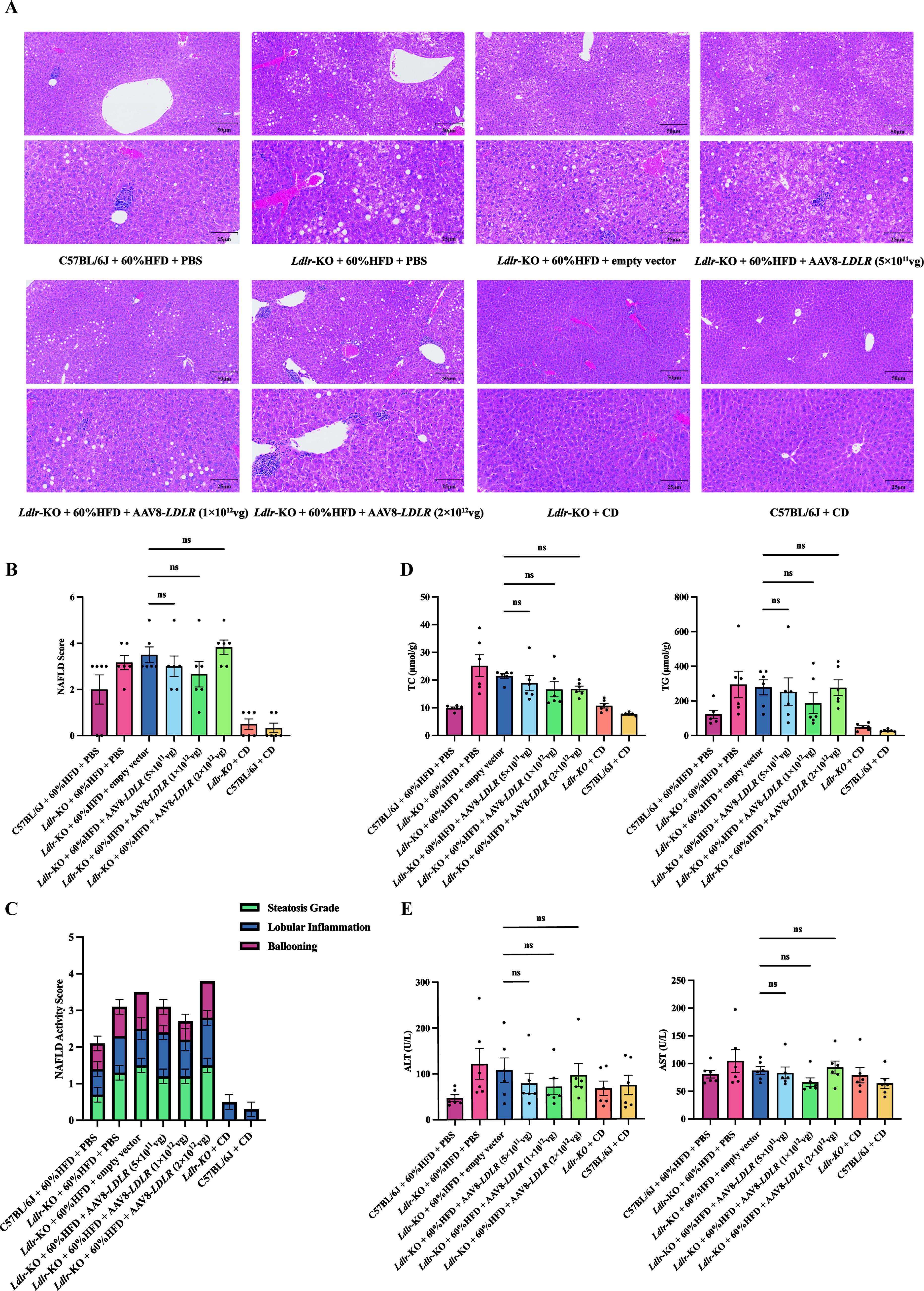
Liver safety and pathology after AAV8-LDLR therapy in Ldlr-KO mice.
A separate assessment for non-alcoholic steatohepatitis (NASH) classified scores of 5 or higher as NASH, scores of 3–4 as suspected NASH, and scores below 3 as non-NASH. Compared to the empty vector control, the proportion of non-NASH cases was significantly higher in the low-dose and medium-dose groups. However, this improvement was not observed in the high-dose group (Fig. 2C and Supplementary Fig. 1A).
To further evaluate potential hepatotoxicity, we measured TC and TG in liver homogenates, along with serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. The TC levels in liver homogenates were lower in the high-dose group compared to the control, but no significant differences were observed between the low-dose and medium-dose groups. TG and aminotransferase levels followed a similar trend to the NAFLD scores, with decreases in the low- and medium-dose groups and increases in the high-dose group. However, these differences were not statistically significant (Fig. 2D, E).
The elevated NAFLD scores and altered lipid levels in the high-dose group may be due to excessive LDLR expression surpassing hepatocyte compensatory capacity or potential hepatotoxicity from high virus particle doses. Overall, these findings suggest that AAV8-LDLR gene therapy is generally safe, with a favorable safety profile for low and medium doses.
Ldlr p.W483X mice show similar lipid changes to Ldlr-KO mice
To address genetic differences between Ldlr-KO mice and clinical HoFH patients, we developed a mouse model with a homozygous Ldlr p.W483X variation. This variation, common in Chinese HoFH patients, reflects the point variations observed in our clinical cohort (Fig. 3A).

Lipid metabolism in the Ldlr p.W483X mouse model.
The dynamic LDL-C levels in Ldlr p.W483X mice were comparable to those in Ldlr-KO mice, both maintaining consistently high levels (Fig. 3B). Western blot analysis confirmed that the LDLR protein expression in the liver of mutant mice was the same as that of Ldlr-KO mice (Fig. 3C). Endpoint lipid analysis further confirmed that Ldlr p.W483X mice exhibited significantly elevated levels of TC, TG, LDL-C, and HDL-C compared to C57BL/6J mice, with no significant difference between Ldlr p.W483X and Ldlr-KO mice (Fig. 3D).
Pathological examination of liver tissues through H&E staining, NAFLD scoring, and analysis of liver homogenate TC and TG levels revealed that Ldlr p.W483X mice displayed similar liver pathology, NAFLD scores, and lipid levels as Ldlr-KO mice (Fig. 4A–C and Supplementary Fig. 1B). These findings indicate that Ldlr p.W483X mice can serve as an effective model for evaluating drug efficacy in HoFH patients, similar to traditional Ldlr-KO mice.

Liver pathology in the Ldlr p.W483X mouse model.
AAV8-LDLR gene therapy effectively and safely reduces lipid levels in Ldlr p.W483X mice
To further assess the efficacy and safety of AAV8-LDLR gene therapy for HoFH, we tested this therapy in a Ldlr p.W483X mouse model. We used 11- to 13-week-old C57BL/6J and Ldlr p.W483X mice, which were fed a 60% HFD for 4 weeks. Mice were then treated with either PBS (Group G1), AAV8 empty vector (Groups G2 and G3), or low-concentration AAV8-LDLR (Group G4) via tail vein injection. Following treatment, the mice continued on HFD for an additional 8 weeks. We evaluated changes in body weight, lipid levels, liver cholesterol (TC), TG, and pathological liver changes at the endpoint (Fig. 5A).
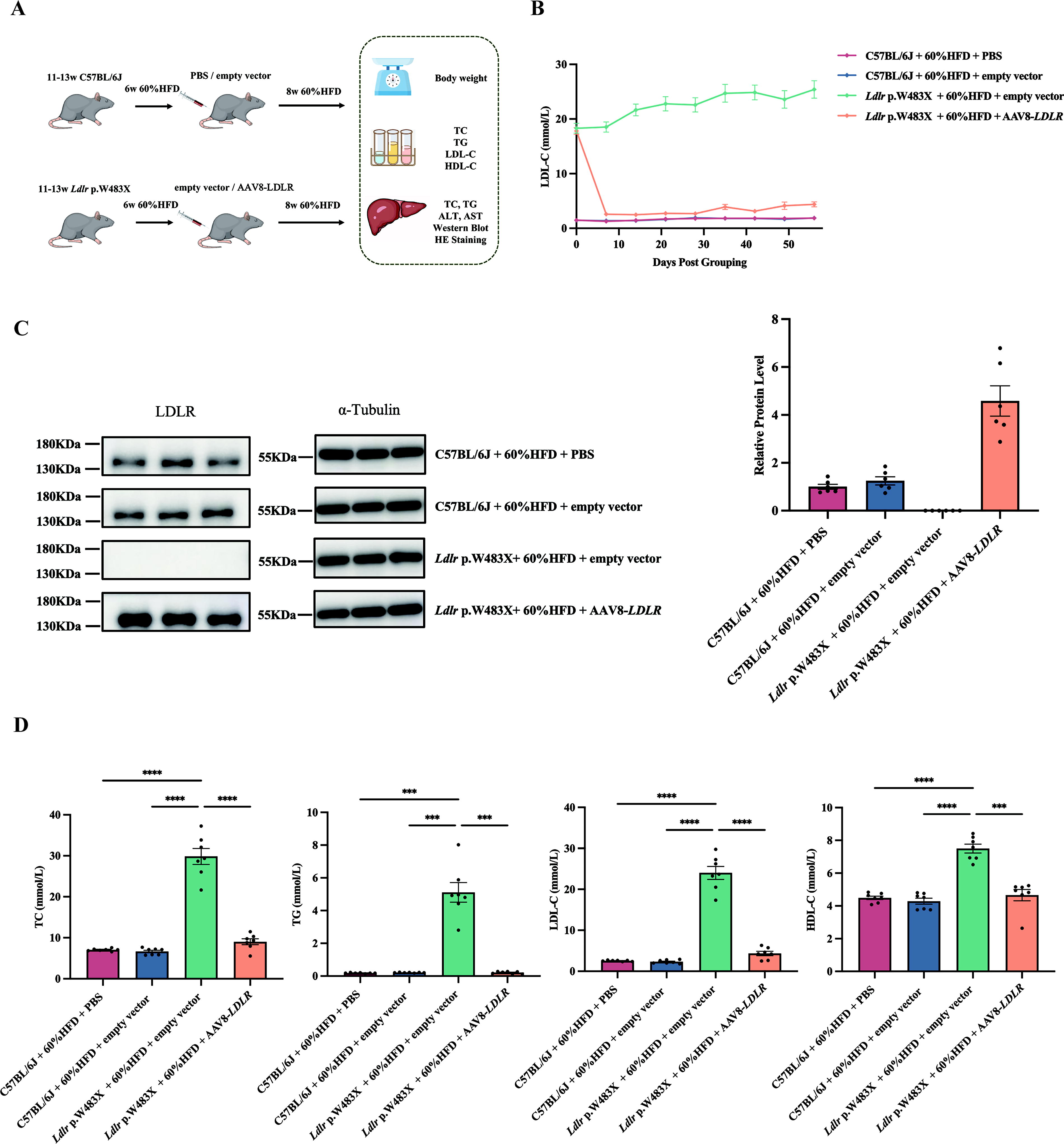
Low-dose AAV8-LDLR therapy attenuates hyperlipidemia in Ldlr p.W483X mice.
Continuous serum LDL-C measurements indicated that mice receiving low-dose AAV8-LDLR gene therapy exhibited a significant reduction in LDL-C levels compared to the control groups, achieving levels close to those of wild-type mice, with the effect maintained over the 8-week period (Fig. 5B). Western blot analysis confirmed that the LDLR protein expression in the liver of mutant mice was restored and treated with a low-dose AAV8-LDLR administration (5 × 1011 vg/mouse) (Fig. 5C). Additionally, AAV8-LDLR gene therapy improved other lipid parameters. At the endpoint, serum levels of TC, TG, LDL-C, and HDL-C in treated mice were similar to those of wild-type mice, showing a significant difference from untreated Ldlr p.W483X mice (p < 0.0001) (Fig. 5D). These results suggest that AAV8-LDLR gene therapy effectively mitigates hyperlipidemia in Ldlr p.W483X mice.
To evaluate safety, we conducted H&E staining on liver tissues and assessed NAFLD scores. There were no significant differences in hepatic steatosis, lobular inflammation, or ballooning across the groups (Fig. 6A). NAFLD scores and NASH ratios also did not differ significantly (Fig. 6B, C and Supplementary Fig. 1C). Although liver TC levels were slightly elevated, TG levels remained consistent (Fig. 6D). The serum ALT and AST levels showed no significant difference across groups (Fig. 6E). These findings indicate that AAV8-LDLR gene therapy does not significantly exacerbate liver burden in Ldlr p.W483X mice. What’s more, to verify whether our gene therapy can effectively prevent the occurrence of AS, we detected the early biomarkers (oxLDL and CRP) that react to AS in the serum of mice by ELISA. The results showed that the levels of oxLDL and CRP in mutant mice that received gene therapy were significantly lower than those in untreated mutant mice. To assess the therapeutic efficacy of AAV8-LDLR in AS prevention, we quantified serum levels of oxLDL and CRP using quantitative ELISA. Comparative analysis revealed that low-dose AAV8-LDLR-treated Ldlr p.W483X mice exhibited reductions in oxLDL and CRP levels, respectively, compared to untreated mutants. Importantly, these biomarkers showed no significant differences between treated mutants and wild-type controls, indicating effective early prevention of AS-associated inflammatory responses (Fig. 6F). Overall, our experiments demonstrate that low-dose AAV8-LDLR gene therapy is both effective and relatively safe for treating hyperlipidemia in this model, suggesting its potential as a future therapeutic option.
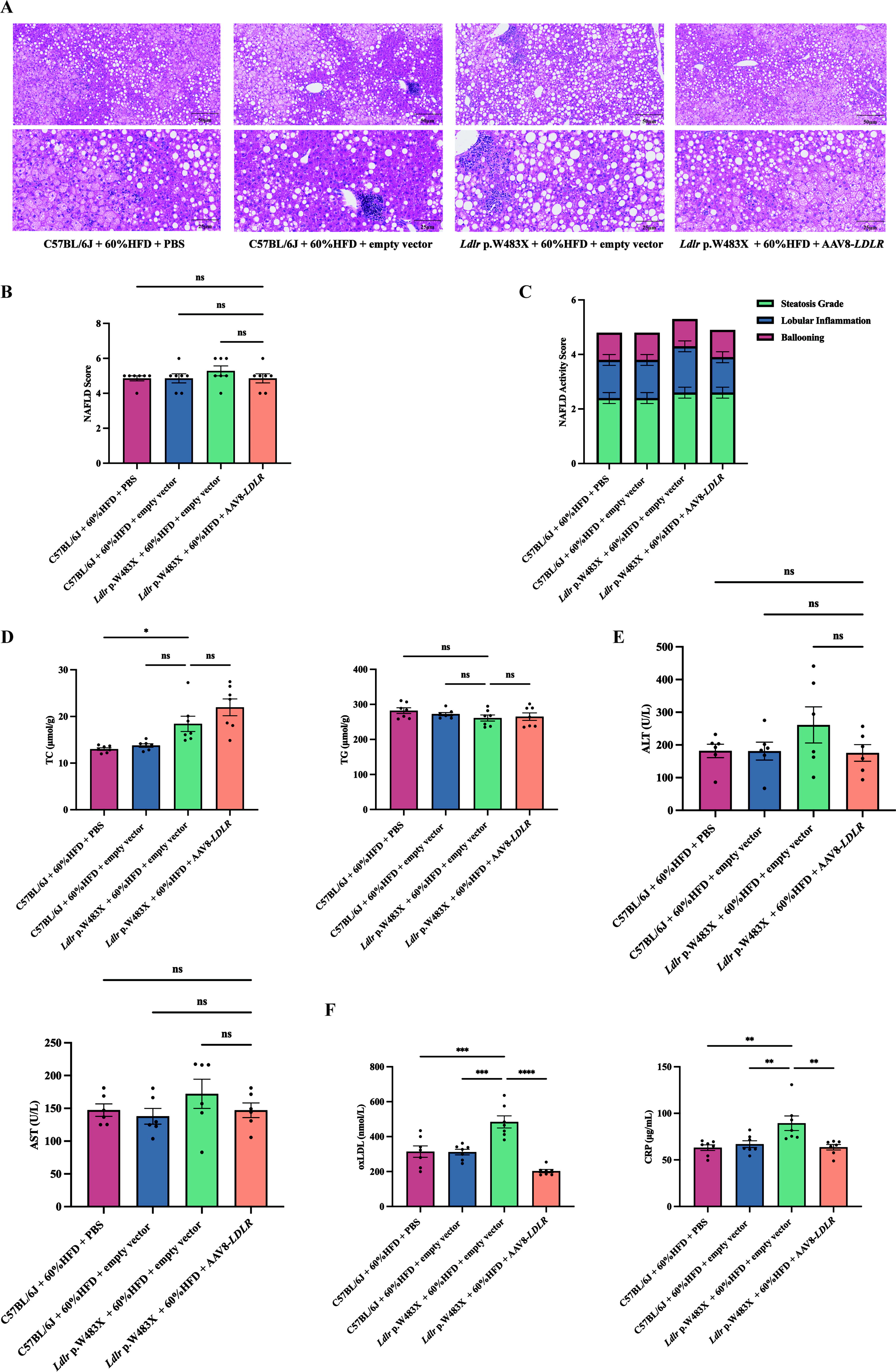
Hepatic outcomes of low-dose AAV8-LDLR therapy in Ldlr p.W483X mice.
DISCUSSION
Recent advances in gene therapy for HoFH have raised concerns regarding safety. In this study, we employed AAV vectors to target both Ldlr-KO and point mutation mouse models, demonstrating substantial restoration of LDLR expression and function. This approach effectively mitigated hyperlipidemia in mice with dose-dependent efficacy. Our results provide strong evidence supporting gene therapy as a promising novel treatment for FH and offer preliminary data on safety, thus laying the foundation for future clinical translation.
Given that most HoFH cases result from loss-of-function mutations in the LDLR gene, which exhibits haploinsufficiency, supplementation of deficient LDLR via gene delivery can effectively improve clinical manifestations. While CRISPR/Cas9-based gene editing faces challenges in efficiently and accurately editing both alleles and raises safety concerns, such as off-target effects, it was not selected for our therapeutic strategy. 19 Current gene delivery vectors are categorized mainly into viral and non-viral systems. Among viral vectors, AAV is more commonly utilized and practical. 20 We chose AAV8, which efficiently targets hepatocytes, as our delivery vehicle and incorporated a TBG promoter to enhance liver-specific expression. 21 Notably, AAV vectors generally do not integrate into the host genome and maintain transgene persistence as episomes, allowing for long-term stable expression in hepatocytes. 22 Although lipid nanoparticle-based non-viral systems also display some hepatocyte specificity, they are susceptible to lysosomal degradation and thus require either high doses or repeated administrations to sustain therapeutic effects. 23
Compared to the phase 1/2 clinical trial by RegenxBio (NCT02651675) utilizing AAV8-mediated LDLR gene transfer, our AAV8-LDLR therapy optimized the dosing regimen. The lowest dose in that trial was 2.5 × 1012 genome copies (gc)/kg but was associated with severe adverse events in two out of three patients, and the trial was halted due to sponsor withdrawal. In contrast, we established 5 × 1011 vg/mouse as a safe and efficacious dose. Translating this dose based on a 42.3-g mouse mean body weight corresponds to 1.18 × 1013 vg/kg. Using body surface area scaling ratios for interspecies dose conversion (mouse:rat = 6.3; mouse:dog = 1.87; mouse:non-human primate = 1.05; mouse:human = 9.1), the equivalent doses are as follows: rats, 1.87 × 1012 vg/kg; dogs, 6.31 × 1012 vg/kg; non-human primates, 1.12 × 1013 vg/kg; humans, 1.3 × 1012 vg/kg. These doses were further adapted considering viral serotype tropism and organ-specific transduction efficiency. 24,25 Furthermore, the p.W483X mutation in LDLR, a recently identified prevalent variant among Chinese FH patients, was incorporated into our mouse model, thereby addressing a gap in FH research relevant to this population. 26
Despite these encouraging results, our study primarily focused on lipid metabolic parameters and liver toxicity. The critical clinical feature of HoFH, AS, was only indirectly assessed by evaluating inflammatory responses and oxLDL levels; direct evaluation of the prevention of atherosclerotic plaque formation was not conducted, possibly due to the relatively short treatment duration insufficient for conventional AS modeling. The genetic heterogeneity of FH, including variations in other genes such as PCSK9 and APOB, should also be considered in future investigations. 27
In conclusion, LDLR gene therapy demonstrates substantial potential as a treatment for FH. Nevertheless, overcoming current challenges and optimizing therapeutic protocols will be essential for successful clinical application. Continued research is warranted to elucidate the mechanisms of action, refine delivery systems, and ensure long-term safety and effectiveness.
Footnotes
ACKNOWLEDGMENTS
The authors thank Biomedical Engineering Facility of National Infrastructures for Translational Medicine, Institute of Clinical Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, and Peking Union Medical College, Beijing 100730, for providing the experimental platform.
AUTHORS’ CONTRIBUTIONS
Q.L. and M.T. contributed equally to this work and shared first authorship. Q.L. and M.T. contributed equally to this work and shared corresponding authorship. X.L. and S.Z. made contributions to the conception and design of the work, Q.L. and M.T. wrote the article. Q.L. and M.T. did mice breeding, blood lipid detection, Western blot, and H&E staining. G.S., Y.L., K.J., and S.Z. helped to construct the vector. X.L., S.Z., and Y.J. were the major contributors in the critical revision of the article. Funding secured by S.Z. All authors read and approved the final article.
AUTHOR DISCLOSURE
The authors have no competing interests to declare.
FUNDING INFORMATION
This work was supported by the
SUPPLEMENTARY MATERIAL
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.