
Abstract
β-thalassemia and sickle cell disease (SCD) are global monogenic blood system disorders, and reactivated δ-globin is expected to replace missing or abnormal β-globin. With the development of gene editing technology, activating γ-globin for treating β-thalassemia and SCD has been highly successful. However, δ-globin, as another important potential therapeutic target, has few related studies. Gene editing technology introduced cis-acting elements, including NF-Y, KLF1, GATA1, and TAL1, into the regulatory region of HBD, successfully activating the expression of δ-globin. It was confirmed that the activation effect of δ-globin was closely related to the location of the introduced cis-acting elements. In this study, the mutation creates a de novo binding site for KLF1 at −85∼93 bp upstream of the transcription start site of the HBD gene, as well as the site for TAL1 and GATA1 cobinding motifs at −59 to ∼−78 bp, which could effectively activate δ-globin.
BACKGROUND
Beta-thalassemia and sickle cell disease (SCD) are the most common inherited blood disorders. β-thalassemia is caused by defects in the HBB gene resulting in insufficient production of β-globin, leading to an imbalance in the ratio of α- to β-globin. In contrast, SCD is caused by point mutations in the HBB gene, leading to the abnormal production of mainly sickle hemoglobin (HbS, α2βS2), which reduces erythrocyte flexibility and damages the cell membrane. In the past, allogeneic hematopoietic stem cell transplantation (HSCT) was thought to be the only practical option to eradicate β-thalassemia and SCD. However, the difficulty of matching and risks such as graft-versus-host disease have limited its widespread use. 1
In recent years, the development of gene therapy and gene editing technologies has provided new ideas for the treatment of β-thalassemia and SCD. Through gene repair or gene activation, 2 it is possible to improve the imbalance of α/β globin chains or replace abnormal β-globin to solve the problems of ineffective hematopoiesis and iron overload caused by the induction of excessive α-globin accumulation and abnormal hemoglobin production. 3,4 A model based on the presence of large numbers of hereditary persistence of fetal hemoglobin (HPFH) in natural populations has led to significant efficacy of activated fetal hemoglobin (α2γ2, HbF) for the treatment of β-thalassemia. 5 –8 The HPFH model has led to the significant success of activated fetal hemoglobin (α2γ2, HbF) in the treatment of β-thalassemia. Theoretically, HbA2 is more advantageous as a replacement for the missing β-globin because the oxyhemoglobin dissociation curve of HbA2 is more similar to HbA. 9,10 However, there are few natural mutations associated with high δ-globin expression in natural populations, so current approaches to activate δ-globin are very limited. Relevant studies have suggested that the creation of KLF1 and NF-Y transcription factor binding sites in the promoter region of HBD plays a role in the activation of HBD. 11 –14 However, this has not been validated in human hematopoietic stem cells, although recent studies have also attempted to validate this in human hematopoietic stem and progenitor cells (HSPCs) using a KLF1 factor binding site introduced into the promoter of the HBD gene, which was almost ineffective in enhancing δ-globin expression. 15 Considering that the location of the transcription factor may have influenced the final results, we attempted to mimic the location of the promoter element of HBB by introducing the KLF1 factor binding sequence CCNCACCCT in the region of −85 bp upstream of the transcription start site (TSS) of the HBD gene and its vicinity. We investigated the effect of creating the KLF1 binding motif at different locations on activating δ-globin.
In addition to KLF1, GATA1 has been recognized as an essential transcription factor in erythroid cell development. 16 –19 Previous studies have successfully used the KLF1-GATA1 protein complex to increase the expression of δ-globin. 20,21 This suggests that GATA1 is also involved in the transcriptional regulation of HBD. There are approximately 3,000 TAL1/GATA1 complex sites existing in the entire genome, consisting of Ebox (CANNTG) or half-Ebox (CTG) motifs located at 7 or 8 bp upstream of the WGATAR motifs, forming CTG-N(7–8)-WGATAR or CANNTG-N(7–8)-WGATAR sequence structure (W denotes T or A base, R denotes A or G base, and N denotes any of A, G, C, or T), which plays an important role in erythropoiesis. 22 Therefore, we attempted to insert the CTG-N (7–8)-WGATAR or CANNTG-N(7–8)-WGATAR) sequence into the non-transcribed region of the HBD and verified whether the sequence structure positively regulates δ-globin expression.
Gene therapy approaches based on autologous HSCT have been investigated as an important therapeutic option for patients lacking a compatible donor for allogeneic HSCT. 23 We hope to develop new potential therapeutic targets for β-thalassemia and SCD by introducing common binding sequences, including NF-Y, KLF1, and GATA1/TAL1, at different sites in the HBD to activate δ-globin through gene editing technology.
RESULTS
Creating a new de novo binding site for NF-Y or KLF1 in the HBD promoter region
For experimental consistency, the CCNCACCCT motif was inserted into the TSS upstream of HBD and its nearby region (Fig. 1a), referring to the promoter region of the HBB gene, which contains a KLF1 binding site at −85 and −100 bp upstream of the TSS. The sequence bases of the inserted KLF1 sites were all CCACACCCT motifs, and the constructed template chains were centered on the insertion sites, with a length of 120 bp single-stranded oligodeoxynucleotide (ssODN), and in the first and last three bases were modified by phosphorylation to ensure the stability of the template strand. As there are few sgRNAs available in the promoter of the HBD we wish to edit, we designed crRNAs to work simultaneously with cpf1 (Cas12a), and to ensure the efficiency of the editing, we chose the sgRNA/crRNA closest to the editing target site and the specific sgRNA/crRNA and template strand used for the different sites. The crRNA and template strand sequences are shown in Supplementary Table S1.
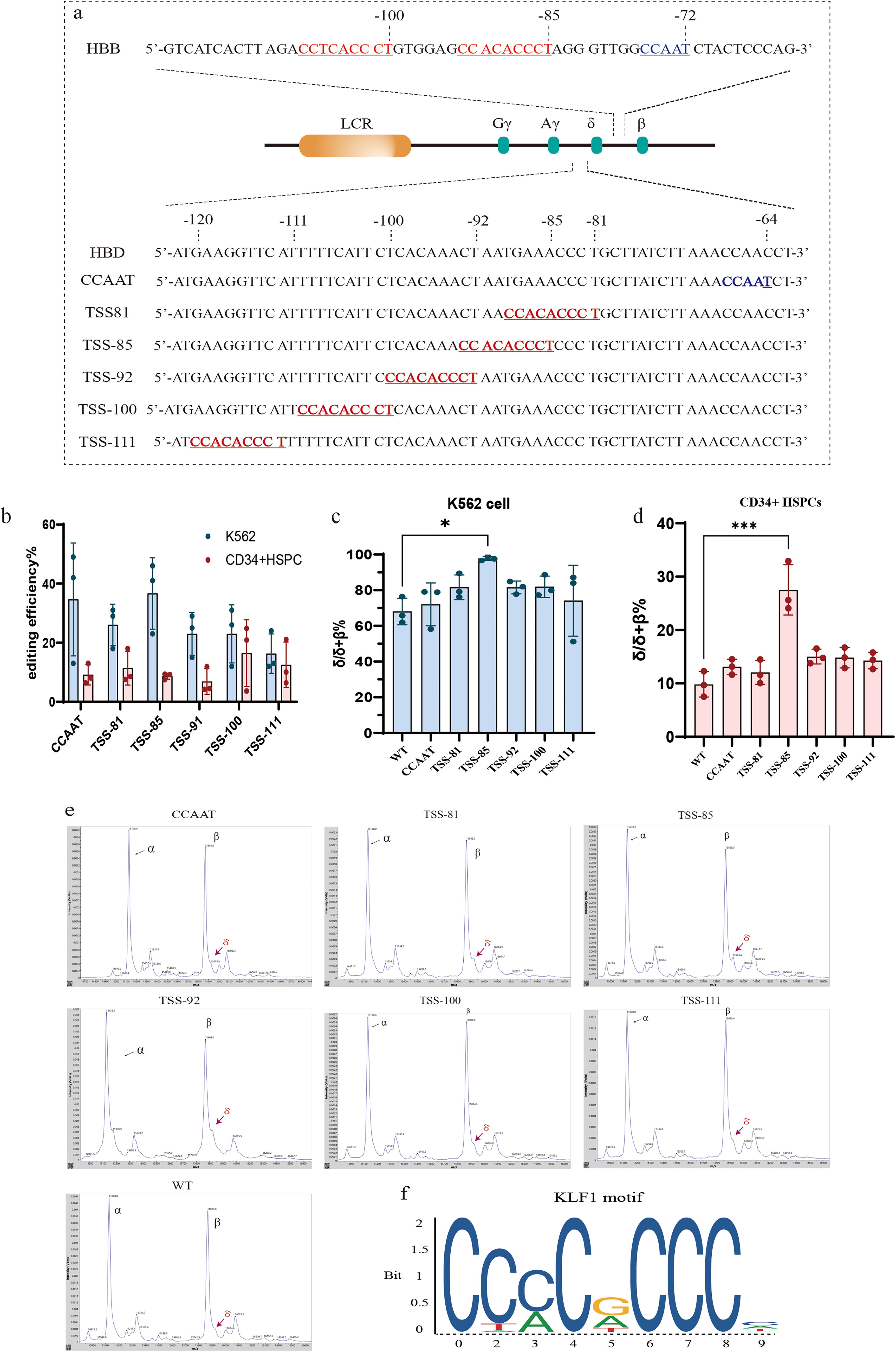
In addition, we created the binding site of NF-Y in HBD, which may be one of the reasons why the expression of HBD was significantly lower than that of HBB, so we simultaneously mutated the sequence of CCAAC to CCAAT, which was located at −64 to −68 bp upstream of HBD (Figure 1a).
We performed electroporation on K562 cells and healthy donor-derived CD34+ HSPCs, respectively, in which the NF-Y factor binding motif was changed from C to T only at −64 bp upstream of the TSS of the HBD gene, whereas the KLF1 binding sequences were introduced at five positions of the HBD gene, including TSS-81 (TSS-81 to −89 bp), TSS-85 (TSS-85 to −93 bp), TSS−92 (TSS-92 to −100 bp), TSS-100 (TSS-100 to −108 bp), and TSS-111bp (TSS-111 to −119 bp), which are the positions to introduce the CCACACCT sequence. The editing efficiency is shown in Figure 1b. The results showed a strong HBD activation effect at the site of TSS-85 in both K562 cells and CD34+ HSPCs. The changes of cell RNA levels before and after editing were evaluated by quantitative PCR with reverse transcription (RT-qPCR), K562 cells increased the ratio of δ/δ + β% at TSS-85 from 68.04% ± 4.30% to 97.73% ± 0.67% (mean ± SEM), and the ratio of δ/δ + β% at other sites was slightly increased, but the effect was not significant(Fig. 1c). In the CD34+ HSPCs, the δ/δ + β% value in the unedited group was 9.83% ± 1.38% (mean ± SEM). A significant increase in the δ/δ + β% value was observed at the TSS-85 site, reaching 27.53% ± 2.73% (Fig. 1d). The results of MALDI-TOF MS confirmed that δ-globin was also significantly elevated at the protein level. Analysis showed the ratio of δ/β was 0.22 ± 0.03 (n = 3, mean ± SEM) at TSS-85 (−85 to ∼−93 bp upstream of the TSS), but it cannot detect the δ-globin in other groups (Fig. 1e). The introduction of CCAAT sequences had little effect on the activation of HBD expression. Insertion of the KLF1 binding motif at the remaining positions resulted in a slight but insignificant activation. This suggests that the location of the transcription factor binding site may be involved in activating δ-globin.
Create the complex binding motif of GATA1 and TAL1 in potential regulatory regions of the HBD gene
Both GATA1 and TAL1 are important transcription factors in erythropoiesis and play important roles in regulating hemoglobin. Some studies identified that TAL1 and GATA1 form a complex at a compound motif consisting of a CANNTG/CTG 7 or 8 bp upstream of a WGATAR motif, which has a more substantial activation effect on the transcriptional regulation of genes. 22 We tried to construct CTG-N(7–8)-WGATAR or CANNTG-N(7–8)-WGATAR sequences through deletion, insertion, or base substitution in different potential regulatory regions of the HBD gene and tested the expression of δ-globin.
In the region of 2,000 bp upstream of the HBD gene, intron, and 3′ flanking region, we found candidate sites that can introduce CTG-N(7–8)-WGATAR or CANNTG-N(7–8)-WGATAR sequence by modifying few numbers of bases, which are T1400 (−1,470 to −1,454 bp upstream of the TSS), T60 (−78 to −59 bp upstream of the TSS), N70 (+214 to +229 bp downstream of the TSS), and N800 (+1,375 to +1,391 bp downstream of the TSS), W60 (+1,703 to +1,719 bp downstream of TSS). We performed electroporation on the K562 cell line and healthy donor-derived CD34+ HSPCs (Figure 2a). Cas9 protein and sgRNA target for the editing site were used to form an RNP complex, and ssODN was added as a homology-directed repair (HDR) template strand.
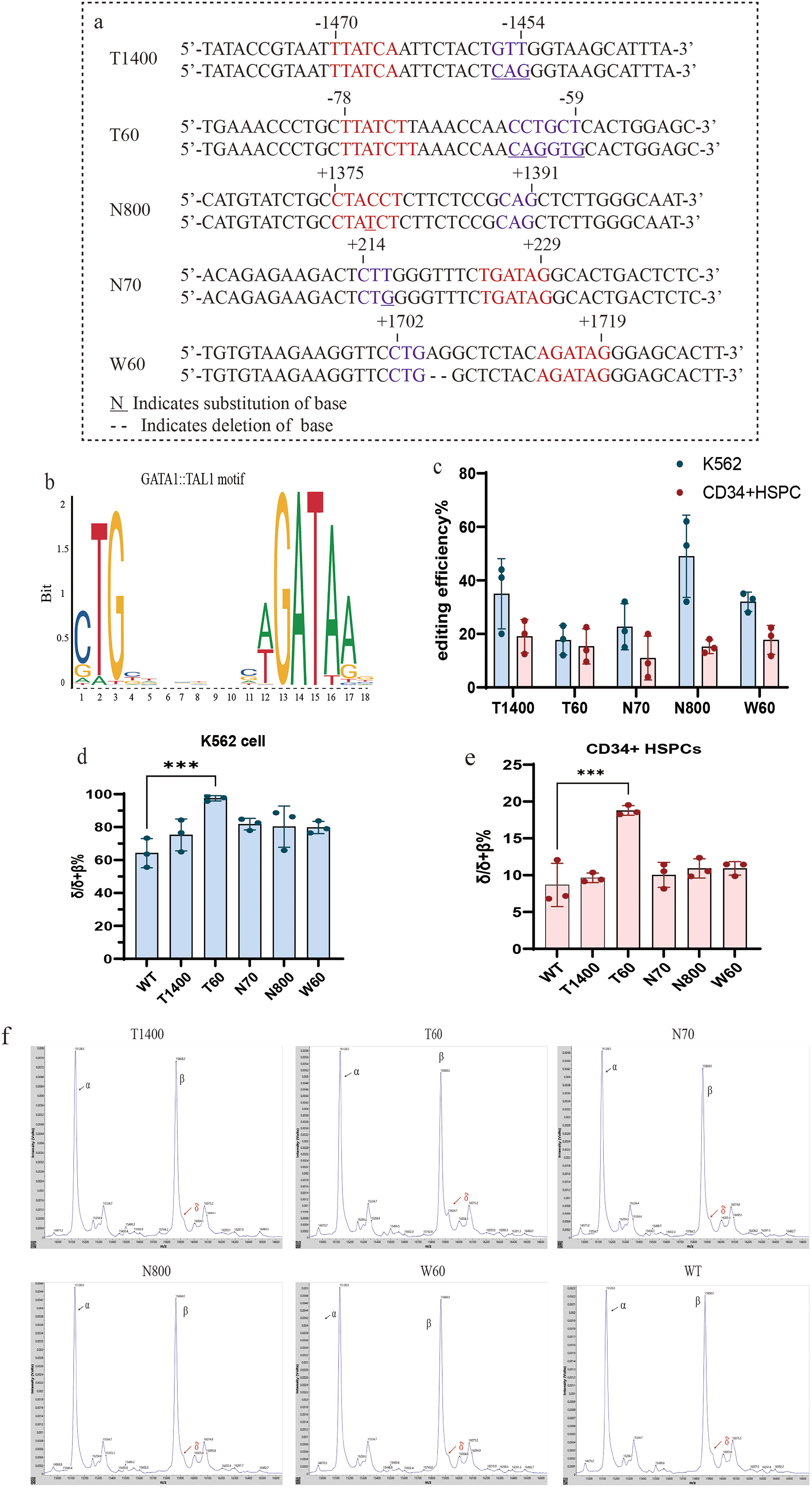
The HDR rates range from 12% to 62% in the K562 cell line and 3.8% to 24.8% in CD34+ HSPCs (Fig. 2c). After editing, the changes of δ-globin at RNA and protein levels were determined. The results indicated a substantial augmentation in the expression of the δ-globin gene at the T60 locus. This augmentation was characterized by an escalation in the δ/δ + β% from 64.17% ± 5.11% to 97.5% ± 0.96%, as observed in K562 cells (Fig. 2d). For CD34+ HSPCs cells, δ/δ + β% was 8.68% ± 1.70% in unedited normal donor-derived CD34+ HSPCs, whereas the result at the T60 locus was 18.79% ± 0.38% (mean ± SEM), showing that δ-globin is also significantly elevated at the RNA level in CD34+ HSPCs(Fig. 2e). MALDI-TOF MS analysis showed the ratio of δ/β was 0.20 ± 0.03(n = 3, mean ± SEM) at the T60 locus, while it cannot detect the δ-globin in other groups (Fig. 2f).
The results suggest that the creation of a common transcription factor binding sequence of GATA1 and TAL1 at the specific locus by gene editing technology, which results in the formation of CTG-N(7–8)-WGATAR or CANNTG-N(7–8)-WGATAR motif in the regulatory region of the HBD gene, can activate δ-globin effectively.
METHODS AND MATERIALS
Cell culture and differentiation
We obtained peripheral blood CD34+ HSPCs after human granulocyte colony-stimulating factor (G-CSF) mobilization from a healthy donor from the Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University (Guangzhou, China), and the study was approved by the Regional Research Review Committee (SYSKY-2024-703-01). The obtained specimens were sorted using CD34 MicroBead Kit (#130-046-702, Miltenyi Biotec), and the sorted CD34+ cells were cultured in X-vivo medium containing 100 ng/mL TPO, 100 ng/ml Flt-3, and 100 ng/mL SCF for 48 h.
Twenty-four hours after electroporation, HSPCs were transferred to erythroid differentiation medium (EDM) consisting of IMDM supplemented with 12 IU/ml erythropoietin (EPO), 5% human AB serum, 10 μg/ml recombinant human insulin, 330 ug/ml holo-human transferrin, 2 IU/ml heparin,1% L-glutamine, and 1% penicillin/streptomycin. During days 0 to 7, EDM was further supplemented with 1 umol/L hydrocortisone (MCE), 100 ng/ml SCF (novoprotein), and 5 ng/ml IL-3 (novoprotein) as EDM-1. During days 7 to 11 of culture, EDM was supplemented with 100 ng/ml human SCF only as EDM-2. No additional supplements were given to EDM as EDM-3 during days 11 to 21 of culture. The culture medium was changed every 3–4 days, and the cell culture density was maintained at 4 × 105/cell to ∼1 × 106 cells/ml.
K562 cells were cultured in 1640 basal medium containing 10% fetal bovine serum and 1% antibiotics and passaged at a density of 0.2–0.5 × 106 cells/ml every 2–3 days.
sgRNA design, preparation of RNP complexes, and cellular electroporation
The sgRNA or crRNA closest to the target sequence was synthesized by screening in the proposed editing region. In total, 250 pmol sgRNA was mixed with 15 ug of spCas9/Cas12a protein and incubated for 20–25 min to form RNP complexes, then 100 pmol of ssODN was added as the template strand (all ssODNs were 120 bp with phosphorothioate modifications in the first and last three nucleotides). Both sgRNA and ssODN were synthesized by Genscript. The number of cells in each group is 4 × 105. After resuspension with PBS, the cells were mixed with the RNP and ssODN complexes and then subjected to electroporation; electroporation was performed using the Lonza 4D Nucleofector (#V4XP-3032, Lonza) for CD34+ HSPCs and the SF Cell Line 4D-Nucleofector™ X Kit S(#V4XC-2032.Lonza) for K562 cells. The program used for electroporating K562 cells was FF120, and CD34+ HSPCs were electroporated using the EO-100 program (Nucleofector 4D).
DNA extraction and editing efficiency testing
Editing efficiency assays were performed 48–72 h after electroporation for K562 cells and CD34+ HSPCs. A total of 1∼5 × 105 cells were harvested, and genomic DNA was extracted using the SteadyPure Universal Genomic DNA Extraction Kit (Accurate Biotech, #AG21010). PCR amplification of the target fragments was performed using Taq polymerase premix (#AG11112, Accurate Biotech). The primers used are listed in Supplementary Table S1, and the PCR program was as follows: 94 degrees for 3 s, 35 cycles of 98 degrees for 10 s, 55 degrees for 30 s, and 72 degrees for 1 min, followed by 72 degrees for 5 min. For the K562 cell line, PCR was performed, followed by Sanger sequencing and EditR analysis. 24 For CD34+ HSPCs, the PCR amplification products were purified using VAHTS DNA Clean Beads (Novozymes, # N411-02) and Illumina-compatible barcoded DNA amplicon libraries were prepared using the SynplSeq DNA Library Prep Kit v1 plus for Illumina (Beijing Xunshi, # XS-L-022–1) and the SynplSeq Universal Adapter System-Plate1 (Twist, # 101308). The products were sequenced on the Illumina Novaseq 6000 system and analyzed using CRISPResso2. 25
RNA extraction, reverse transcription, qRT-PCR
The K562 cells were electroporated and subsequently cultured for seven days. The CD34+ HSPCs cells were then induced to differentiate for 14 days. A total of 1 × 106 cells were harvested, and total RNA was extracted using the SteadyPure Universal RNA Extraction Kit (Accurate Biotech, #AG21017). RNA isolation and reverse transcription were performed using the Evo M-MLV RT Mix Kit (Accurate Biotech, #AG11728). RNA was then subjected to gDNA removal and reverse transcription. Real-time fluorescent quantitative polymerase chain reaction (qPCR) was performed using the SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biotech, #AG11718). DNA samples from the CDS region of the HBD and HBB genes were utilized as the standard to construct a standard curve. The gene copy number of the HBD and HBB genes was then calculated by determining the CT value of the samples. The final result was presented as the ratio of the HBD gene copy number to the HBD plus HBB copy number (δ/δ + β%). The primers used are listed in Supplementary Table S1.
MALDI-TOF MS analysis
CD34+ HSPCs were detected by Matrix-Assisted Laser Desorption/Ionization time-of-flight mass spectrometry (MALDI-TOF MS) on day 14 of differentiation using 3∼4 × 106 cells taken from each group. 26 Each 2× 105 cell was diluted and mixed with 50 μl of ultrapure water, and the diluted samples were mixed with matrix solution (10 mg/ml sinapinic acid [Sigma-Aldrich, St. Louis, USA], 40% CH3CN, and 0.1% TFA) in a 1:9 ratio. 2.5 μl of the mixture was dispensed onto a stainless steel MALDI target plate, which was kept warm with a metal bath during the dispensing process (the temperature was set at 39°C) and then dried and examined on a machine (QUAN-TOF, Intelligene Biosystems, Qingdao, China). The parameters were as follows: laser pulse energy was 4.8 μJ; m/z range was 5,000–20,000; acceleration voltage was 20 kV; scan rate was 0.5 mm/s; and ten rows were scanned per sample spot; laser pulse frequency was 1 kHz; focus mass was 15,000. Each sample was subjected to a minimum of 30 individual spectra (800 shots per spectrum), comprising over 20,000 effective laser shots.
Statistical analysis
Data were analyzed by unpaired two-tailed Student’s t-test and presented as mean ± SEM (Standard error). A value of p < 0.05 was considered to be a statistically significant result. Data were also analyzed and plotted using GraphPad Prism version 10.0.
DISCUSSION
Current gene therapy options for β-thalassemia and SCD include repairing the missing or mutated HBB gene or activating other β-like-globin (γ, δ) to replace missing or abnormal β-globin. In this study, the important finding is that the expression of δ-globin is elevated by introducing binding motifs for KLF1, GATA1, and TAL1 at specific sites of −85 to ∼−93 bp and −59 to ∼−78 bp upstream of the TSS of the HBD gene, which can be potentially used to replace the missing or abnormal β-globin to treat β-thalassemia and SCD.
In theory, δ-globin should have an advantage over γ-globin as it is a type of β-like globin that is expressed by HBD, is highly homologous and conserved with HBB, and is more widely distributed in cells. 27 Previous studies have attempted to increase HbA2 (α2δ2) in SCD mice and found that HbA2 effectively ameliorated the symptoms associated with SCD and β-thalassemia mice. 28,29 This suggests that δ-globin may resist a number of pathophysiological abnormalities caused by the absence or abnormality of β-globin.
However, there is limited information on the means of activating δ-globin, and few individuals with congenitally high δ-globin have been identified in the natural population individuals. For γ-globin activation, the leading genetic options are to create a new transcription factor binding site in the regulatory region of the gene or to disrupt the binding site of a known repressor element (e.g., BCL11A, ZBTB7A). 8,30,31 Unlike γ-globin, δ-globin has never dominated the developmental transition of human globin, and no critical repressor elements have been identified. Therefore, creating a new transcription factor binding site in the HBD regulatory region is a worthwhile activation scheme. In the past, there have been cases where HBD expression has been increased by introducing mutations in K562 and MEL cell lines, but the practice in HSCTs is lacking. At the same time, some researchers have recently found that the insertion of a binding site for KLF1 upstream of the TSS in HBD by gene editing technology had no significant effect on the elevation of δ-globin, 15 which is contrary to previous research using transgenic technology. 11,12
To further explore the gene editing scheme for effective activation of δ-globin, we modeled the KLF1 binding sequence by introducing the CCNCACCCT sequence at the location of the HBB (at −85 and −100 bp upstream of the TSS) as well as in other regions in the vicinity of the KLF1 binding site. We confirmed that δ-globin expression can be efficiently activated by inserting the KLF1 binding site into the promoter region of the HBD and that the activation effect correlates with the position of the KLF1 binding site. The activation effect is related to the position of insertion, and the best effect is found at TSS-85 (−85 to −93 bp upstream of TSS), which is the same as the sequence position of the KLF1 binding site of the HBB gene. We also corrected CCAAC to CCAAT in the HBD promoter region to create the NF-Y binding site. However, this did not seem to have a significant effect on the activation of HBD expression. Previous studies have sought to examine the impact of NF-Y on gamma-globin expression. In contrast to the conclusions drawn in this study, the aforementioned research suggests that NF-Y plays a significant regulatory role in this process. 32 However, we did not attempt to introduce the binding sequences of NF-Y in different regions for comparison, so further validation is required.
GATA1 and TAL1 are also members of the erythroid essential transcription factors. Creating mutations in the noncoding region of the human globin gene to generate new binding site structures for GATA1 or TAL1 factors can recruit transcriptional activators such as GATA1/TAL1 to promote the expression of the target globin during cell differentiation into erythroid cells. 33 –35 In contrast, the disruption of GATA1 or TAL1 binding sequences in genes has an impact on erythropoiesis. 4,36 Although thousands of GATA sequence structures exist in the human genome, not every GATA motif has a positive regulatory element. One such study has demonstrated that the site where GATA1 acts as an activator usually coexists with a TAL1 binding sequence (CANNTG). In contrast, at sites where the TAL1 sequence is absent, GATA1 is usually an inhibitory factor. 37,38 Thus, the generation of GATA1 and TAL1 cobinding sequences has the potential to reactivate silenced globin genes.
We inserted the GATA1 and TAL1 compound motif by constructing the sequence structure of GATA1 (WGATAR) and its upstream 7–8 bp containing NTG or CANNTG. The noncoding region of the HBD gene was screened in the hope that a small number of bases could be modified to form the CTG-N(7–8)-WGATAR or CANNTG-N(7–8)-WGATAR motif. By trying in the region of the HBD gene upstream of the TSS, intron, and 3′ flanking, it was possible to react with the δ-globin in the T60 locus (−59 to −78 bp upstream of the TSS of the HBD gene). Importantly, the T60 locus carries a GATA motif, which has been shown to positively regulate δ-globin expression. 39 We further enhanced δ-globin expression by introducing an Ebox element (CANNTG) upstream of the original GATA sequence. The results provide new ideas for globin activation strategies.
The shortcoming of this study is that the repair efficiency of HDR is not high. As the editing efficiency of CRISPR-Cas9/Cas12a in the form of HDR is further reduced in vivo, we did not perform animal experiments. However, with the development of gene editing technology, new gene editing tools, such as base editing and prime editing, are expected to solve the problem of inefficient repair. 8,40,41
In this study, we identified potential activation mechanisms of δ-globin, including important expressed transcription factors involved in δ-globin and critical sites (−85 to −93 bp and −59 to −78 bp upstream of the TSS of the HBD gene) where expression can be activated. This provided a new scheme for the subsequent gene therapy strategy of β-thalassemia and SCD.
Footnotes
ACKNOWLEDGMENTS
The authors thank laboratory members for helpful discussions and support; Chuanli Lu for help with MALDI-TOF MS; Reforgene Medicine for their contribution to the K562 cell line; and all the HSPC donors from Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University who generously participated in this study.
AUTHORS’ CONTRIBUTIONS
L.C. designed and performed the experiments, analyzed the data, and wrote the article. D.L. performed the experiments and analyzed the data. W.H. and L.X. performed the experiments and analyzed the data. L.C., Y.L., H.X., and J.L. provided technical assistance and experimental design. X. L. and J.F. conceived the study, designed the experiments, analyzed the data, and wrote the article. X.L. and J.F. contributed equally to this study.
AVAILABILITY OF DATA AND MATERIALS
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
AUTHOR DISCLOSURE
The authors have declared that there are no conflicts of interest. No financial or nonfinancial benefits have been received or will be received from any party directly or indirectly related to the subject of this article.
FUNDING INFORMATION
The work has been supported by the Science and Technology Project of Guangzhou (No. 202201010962) and Guangdong Provincial Key R&D Program (2023B1111050002), the Science and Technology Project of Sun Yat-sen Memorial Hospital (No. YXQH201913), Natural Science Foundation of Guangdong Province (No. 2025A1515012539) and grants for a Clinical Key Discipline (The Subtropical Disease Center for Thalassemia) from the Chinese Ministry of Health (No. 1311200006107).
SUPPLEMENTARY MATERIALS
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.