
Abstract
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). While gene therapy holds promise as a cure, the cell-type-specific heterogeneity of CFTR expression in the lung presents significant challenges. Current CF ferret models closely replicate the human disease phenotype but have limitations in studying functional complementation through cell-type-specific CFTR restoration. To address this, we developed a new transgenic ferret line, CFTR int1-eGFP(lsl), in which a Cre-recombinase (Cre)-excisable enhanced fluorescent protein (eGFP) reporter cassette is knocked in (KI) to intron 1 of the CFTR locus. Breeding this reporter line with CFTR G551D CF ferret resulted in a novel CF model, CFTR int1-eGFP(lsl)/G551D, with disease onset manageable via the administration of CFTR modulator VX770. In this study, we confirmed two key properties of the CFTR int1-eGFP(lsl)/G551D CF ferrets: (1) cell-type-specific expression of the CFTR(N-24)-eGFP fusion protein, driven by the intrinsic CFTR promoter, in polarized epithelial cultures and selected tissues, and (2) functional reversion of the KI allele via Cre-mediated excision of the reporter cassette. This model provides a valuable tool for studying the effects of targeted CFTR reactivation in a cell-type-specific manner, which is crucial for enhancing our understanding of CFTR’s roles in modulating airway clearance and innate immunity, and for identifying relevant cellular targets for CF gene therapy.
INTRODUCTION
Cystic fibrosis (CF) is a recessive, multiorgan, life-threatening disease caused by a defect in a single gene: cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is a cyclic adenosine monophosphate (cAMP)-dependent ion channel protein that facilitates the transepithelial transports of chloride (Cl−) and bicarbonate (HCO3−) ions. 1,2 While CF affects multiple organs, the main cause of death is chronic bacterial infection in the lungs caused by dehydrated mucus that impairs the clearance and killing of inhaled pathogens. In CF lungs, dysfunction of the CFTR channel results in inadequate hydration and a thickened mucous layer. Consequently, mucociliary clearance is impaired, resulting in defective innate immunity and spontaneous bacterial colonization that can ultimately cause pulmonary failure. 3 Within the respiratory system, each cell type has its own unique combination of expressed genes that impart distinct cellular functions, and these functions across multiple cell types coordinate airway surface liquid hydration, mucociliary clearance, and innate immunity in the airway. 4 Gene therapies for CF lung disease are an attractive treatment approach due to their potential to permanently restore CFTR function, regardless of patient’s genotype, and are therefore considered a promising path toward an ultimate cure for the disease. 5,6 As CFTR expression is tightly regulated in a cell-type-specific manner along the respiratory tree, 7,8 effective gene therapy may require the restoration of regulatable CFTR expression in multiple cell types to rebuild normal airway homeostasis. 9 However, the complexities of CFTR biology and function in the respiratory system are still not fully understood. As a result, the specific cell types that require CFTR complementation to prevent or reserve disease remain largely unknown.
Animal models that recapitulate human disease phenotypes and progression are crucial for advancing our understanding of disease mechanisms, facilitating drug discovery, and testing new treatments, such as gene therapy. 9 In 2006, our group pioneered cloning domestic ferrets (Mustela putorius furo) via somatic cell nuclear transfer. 10 Subsequently, we utilized this technique to produce the first CF ferret, CFTR KO, through CFTR gene knock-out (KO) by disrupting its coding sequence in exon 11. 11 More recently, we used advanced CRISPR/Cas9 genome editing techniques in ferret zygotes to develop ferrets bearing the delF508 or G551D mutations in CFTR. 12 –14 We also established a CFTR conditional KO ferret model (CFTR L/L) to investigate the function of pulmonary ionocytes and understand the consequences of ionocyte-specific CFTR deletion. By breeding the Cre-recombinase (Cre) driver line (FOXI1-CreERT2 ) with CFTR L/L ferrets, which carry a Cre-excisable CFTR exon 16, we generated ferrets in which CFTR expression can be selectively depleted in ionocytes. Using this model, we characterized the functions of ionocytes in anion secretion, absorption, and mucociliary clearance using precise and cell-type-specific CFTR gene deletion. 15 Although existing CF ferret models provide invaluable insights in the pathogenesis of CF and allow for the evaluation of cell-specific CFTR loss-of-function involvement in lung functions, they do not effectively model cell-type-specific functional complementation following gene editing to identify the relevant cellular targets for effective CF gene therapy.
To address this and fully characterize the heterogeneity of CFTR expression in the airway epithelium, we developed a novel CFTRint1-eGFP(lsl) reporter ferret model. In this model, the CFTR gene is disrupted by inserting the splice acceptor-associated eGFP cDNA in the context of a LoxP-STOP-LoxP (lsl) sequence into CFTR intron 1. This insertion leads to the expression of a fusion protein CFTR(N24)-eGFP, hereafter referred to as the eGFP reporter, that merges eGFP with 24 amino acids of the N-terminal CFTR protein (encoded in exon 1 and 5′ end of exon 2). As an exon-trap insertion, the knock-in (KI) of the eGFP reporter hijacks the endogenous CFTR promoter and thus inactivates this CFTR allele; however, CFTR expression can be restored by Cre-mediated excision of the lsl sequence from the KI allele. Therefore, this model can be used to study the consequences of restoring CFTR function in the context of CF, using viral vector-delivered CRE expression or breeding with cell-type-specific Cre driver animals.
We subsequently crossed our eGFP reporter line with the CFTR G551D line 13 to generate a novel CF ferret model, CFTR int1-eGFP(lsl)/G551D. Unlike CFTR-KO ferrets, 16 the gating function of CFTRG551D mutant can be rescued by the CFTR potentiator, VX-770, making the onset of CF disease in CFTR G551D ferrets manageable. Administration of VX-770 during and after the gestation period enables normal growth and survival at birth; however, discontinuing treatment at any age leads to reinitiation of CF disease in the pancreas, gut, and lungs. 14 This alleviates the challenges associated with rearing severely ill animals, making the CFTR G551D CF ferrets particularly suitable for preclinical studies of CF gene therapy. 17,18 In this report, we confirmed two key properties of the CFTR int1-eGFP(lsl)/G551D CF ferrets: the cell-type-specific coexpression of the eGFP reporter and CFTR in polarized airway epithelial cultures in vitro and selected tissues/organs in vivo, and the functional reversion of the KI allele via Cre-mediated excision of the reporter cassette.
METHODS AND MATERIALS
Animal study
This study has been approved by the Institutional Animal Care and Use Committees of the University of Iowa. Due to the limited availability and difficulty in obtaining an equal distribution of genders when using transgenic ferrets, sex was not considered a variable during the transgenic line expansion and characterization.
Construction and production of donor DNA for homology-independent targeted integration
The Cre-responsive eGFP reporter cassette, flanked with a pair of LoxP sites, was assembled using a DNA synthesis service provided by Genscript Biotech (Piscataway NJ). This 3.4 kb sequence includes a 229 base pair (bp) sequence encompassing the splice acceptor site at the intron 1/exon 2 junction of CFTR, the eGFP cDNA, which was fused in-frame to the first 22 bp of exon 2, 1.67 kb 3′ untranslated region (UTR) from the last exon (exon 27) of the CFTR gene to preserve full posttranscriptional regulation for reporter expression, and the polyadenylation (polyA) sequence of the human β-globin gene (hBGpA) as an add-on to strengthen transcription termination. For the production of the plasmid backbone-free minicircle (mc) DNA. 19 as the donor for the homology-independent targeted integration (HITI) approach, 20 the assembled reporter sequence was subcloned into the mcDNA parental plasmid pMC.BESPX-MCS1 (System Bioscience, Palo Alto, CA) between attP and attL sites of the phiC31 integrase. To facilitate “cut and paste” genome manipulation via HITI, a 23 bp sequence, ggaacctagtgaagcta//gtaTGG (the HITI targeting site, recognized by guide (g)RNA3, “//” marked the cleavage site; upper cases represent the protospacer adjacent motif), was incorporated in the opposite orientation as it presents in intron 1. System Bioscience used the resultant producer plasmid, pMC-(sa-eGFP/UTR), for mcDNA production. Ferret CFTR locus spans 169.4 kb and consists of 27 exons (ENSMPUG00000007138, scaffold GL896904.1: 20,224,011–20,393,397, reverse strand). Notably, intron 1 is 29,285 bp in length. The gRNA3 recognition sequence is located at the 3′ end of intron 1 with the CRISPR cut site at 1,056 bp upstream of the exon 2.
Generation of the CFTRint1-eGFP(lsl) reporter ferret line
The founder CFTR int1-eGFP(lsl) ferret was generated using CRISPR-mediated HITI in zygotes to knock in the reporter cassette into the predetermined target site at intron 1 of CFTR, recognized by gRNA3. The collection of fertilized ferret single-cell embryos (zygotes) and zygote microinjection were conducted as previously described. 15 The mix of ribonucleoprotein, spCas9 complexed with gRNA3, and mcDNA donor was microinjected into zygotes (3∼5 picoliters). Injected zygotes were cultured in TCM-199 medium supplemented with 10% fetal calf serum (FCS) overnight to the 2-cell stage before being transferred into primipara pseudo-pregnant jills. The kits were naturally delivered in 42 days (full-term gestation). The gRNA3 was designed using the online tool CRISPOR (http://crispor.tefor.net). spCas9 protein (from Streptococcus pyogenes), gRNA3, and PCR primers for genotyping and TaqMan quantitative PCR assays were generated at Integrated DNA Technologies, Inc. (Coralville, IA). The founder animal was identified by PCR-based genotypic analysis followed by confirmation by Sanger sequencing. Primer sets, L-Fw/int-Rev (L-Fw: caactgtgtatctcttccaaactct; int-Rev: tttacgtcgccgtccagctcga) and int-Fw/R-Rev (int-Fw: cgctttcttgctgtccaatttc; R-Rev: agggctgaatccagtctctatc), amplified the junction sequences at the 5′ (left) and 3′ (right) of the insert, respectively.
Culture and expansion of ferret airway basal cells
Primary tracheal cells and nasal epithelial cells were dissociated and collected from the trachea and nasal turbinate of CFTR int1-eGFP(lsl)/G551D ferrets (3 months old), as previously described. 13 Airway basal cells were expanded in PneumaCultTM-Ex Plus medium (StemCell Technologies, Vancouver, Canada). Trachea biopsy of the founder AA2 was collected with a bronchoscope, as we previously described. 21 Airway basal cells isolated from trachea biopsies were produced following Liu and Schlegel’s method. 22 In brief, epithelial cells dissociated from the biopsy were first cocultured with Cs137-irradiated (3,000 rad) Swiss 3T3-J2 fibroblasts in F-medium (3:1 (v/v) F-12 Nutrient Mixture (Ham)–Dulbecco’s modified Eagle’s medium (Invitrogen), 5% fetal bovine serum, 0.4 g/mL hydrocortisone (Sigma-Aldrich, St. Louis, USA), 5 g/mL insulin (INS) (Sigma-Aldrich), 8.4 ng/mL cholera toxin (Sigma-Aldrich), 10 ng/mL epidermal growth factor (Invitrogen), and 24 g/mL adenine (Sigma-Aldrich) with addition of 5 mol/L Rho-associated kinase (ROCK) inhibitor (Y-27632; Enzo Life Sciences, Farmingdale, NY). After the epithelial colonies were sufficiently expanded (e.g., were visible), they were collected and further cultured in serum-free PneumaCultTM-Ex Plus medium (StemCell Technologies, Vancouver, BC, Canada). The transduction of basal cells with a Cre-expression lentiviral vector HIVUbCpuroPGKCre (VVC-U of Iowa-7555, Vector Core, University of Iowa), and selection for puromycin (Puro)-resistant cell pool was conducted as previously described. 13
Polarized ferret airway epithelial cultures
Airway basal cells were seeded onto 6.5-mm polyester Transwell® inserts (Corning, Glendale, AZ) precoated with collagen IV (Sigma-Aldrich) at a density of 1.5 × 105 cells per insert. Seeding occurred in PneumaCultTM-Ex Plus medium (StemCell Technologies, Vancouver, BC, Canada). At 24 h postseeding, the medium was replaced with PneumaCultTM ALI medium (StemCell Technologies) in both apical and basal chambers. Cultures were then air-lifted the following day to facilitate differentiation of the polarized ferret airway epithelium at an air-liquid interface (ALI) for at least 3 weeks. The basal chamber medium was replaced twice a week during the culture period. Matured ferret airway epithelium-ALI cultures (transepithelial electric resistance >1,000 Ω.cm2) were used for Ussing chamber assays and TaqMan PCR for mRNA assays.
Southern blotting
Following restriction enzyme digestion, ferret genomic DNA was resolved on a 1% agarose gel and transferred to a positively charged Nylon transfer membrane (GE HealthCare, Chicago, IL) for blotting. Target bands were visualized by autoradiography with radioactive phosphorus-32 (32P)-labeled probes, as previously described. 11
mRNA quantification
Total ferret RNA was extracted using Qiagen RNeasy Plus Mini Kit (Qiagen, Germantown, MD) from ALI cultures and converted to cDNA using the ABI High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA) for quantitative RT-PCR (qRT-PCR). Before reverse transcriptase reaction, RNA samples were digested with Turbo DNA-freeTM kit (ThermoFisher Scientific, Waltham, MA) to further decontaminate genomic DNA. The sequences of the primer set and the probe specific for a 124 bp amplicon of the ferret CFTR were as follows: Forward: TGGCTTGGAAATCAGTGAGG (exon 14), Reverse: CTTGTGGATAGTAACATATCGGAGG (exon 15), and Probe: 6FAM-ACTGGTGGTATGCTTTCAACATCGTCA (exon 15). The sequences of the primer set and the probe specific for a 113 bp amplicon of the eGFP were as follows: Forward: GTGAACCGCATCGAGCTGAA, Reverse: TGCTTGTCGGCCATGATATAG, and probe: 6FAM-ATCGACTTCAAGGAGGACGGCAAC. The sequences of the primer set and the probe specific for a 137 bp amplicon of ferret glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were as follows: Forward: CAACTTTGGCATTGTGGAGG, Reverse: CAGTGGAAGCAGGGATGATG, and Probe: 6FAM-CAGTGATGGCATGGACGGTGG. Standard curves were generated using known amounts of plasmids harboring the corresponding DNA fragments to calculate copy numbers.
Short-circuit current measurements in Ussing chambers
The ALI culture inserts were placed under a multichannel voltage current clamp in self-contained P2300 Ussing chambers (Physiologic Instruments, San Diego, CA, USA) for Isc measurement using an asymmetric chloride buffer system, as previously described. 13,15 The following antagonists and agonists were sequentially added to the apical chamber and the changes in current were recorded during the experiment: 1) 100 μM amiloride, 2) 100 μM 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS), 3) 10 μM forskolin (Forsk)/100 μM 3-Isobutyl-1-methylxanthine (IBMX), and 4) 50 μM GlyH-101.
Immunofluorescent analysis
The ferret trachea, nasal turbinates, and pancreas were dissected and fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) (pH 7.4) overnight. They were then processed for whole mount immunostaining. The tissues underwent antigen retrieval by incubating in 10 mM citrate buffer at 55°C overnight on a rocker. Afterward, tissues were washed three times for 30 min each and then blocked with 20% donkey serum/PBS for 6–8 h at 37°C on a rocker. Primary antibodies were applied and incubated at 37°C on a rocker for 2 days. Following this, the tissues were washed three times for 30 min each and then incubated with secondary antibodies for 1 day at 37°C on a rocker. The following antibodies were used: anti-Barttin CLCNK Type Accessory Subunit Beta (BSND) (1/300–1/500; ab196017, Abcam, Waltham, MA), anti-ATP6V1G3 (1/300–1/500; HPA028701, Sigma-Aldrich), anti-CFTR (1/100–1/300, CFTR antibody 596, cftrantibodies.web.unc.edu), anti-Muc5B (1/300, HPA008246, Sigma-Aldrich), anti-FOXJ1 (1/300, ab235445, Abcam), anti-SOX9 (1/300, AB5535, Millipore Sigma), anti-INS (1/300, P01315-INS, Dako), Alexa Fluor 488 donkey anti-chicken IgG (1/400, 703-546-155, Jackson ImmunoResearch, West Grove, PA), Alexa Fluor 555 donkey anti-mouse IgG (1/400, A31570, Life Technologies, Carlsbad, CA), and Alexa Fluor 647 donkey anti-mouse IgG (1/400, A31571, Molecular Probes, Eugene, OR). After incubating with the secondary antibody, the samples were washed three times for 30 min each in PBS, and then transferred to Ce3D tissue clearing solution (Biolegend catalogue no. 427704, San Diego, CA) for 2–3 h at room temperature. Following tissue clearing, the samples were mounted onto Superfrost Plus microscope slides (Fisherbrand, Waltham, MA) under 0.33 mm coverslips, and the edges were sealed with Gorilla Glue. Binder clips were used to clamp the coverslips for 30 min to ensure proper glue fixation. Imaging acquisition was performed using Zeiss LSM 880 or 980 confocal microscopes.
Statistical analysis
All measurements were taken from individual samples. The datasets were analyzed and presented using GraphPad Prism software v8.4. The differences between groups were assessed using a two-tailed Student’s t-test (for paired eGFP and CFTR mRNA assays from each cell lysate, n = 6). Statistical significance was determined, with p < 0.05 being significant.
RESULTS
Generation of the CFTRint1-eGFP(lsl) reporter ferret line
To generate a ferret model that allows cell-type-specific reactivation of CFTR, we inserted a 3.4 kb reporter cassette of splicing acceptor associated-eGFP coding sequence and polyA signals into intron 1 of the CFTR gene to highjack the endogenous promoter via exon trapping (Fig. 1A). The polyA signals include the UTR of the last CFTR exon (exon 27) to preserve the posttranscriptional regulation in this region and the hBGpA from the human β-globin gene to enhance transcription termination. The targeted site for insertion was the gRNA3 recognition sequence within the ferret CFTR intron 1 (1,056 bp upstream of the exon 2). Inserting this sequence at the desired site disrupts the expression of CFTR from the KI allele, resulting in the expression of eGFP reporter [CFTR(N24)-eGFP fusion protein] driven by the endogenous CFTR promoter. Flanking the inserted reporter cassette are two LoxP sites that allow the cassette to be excised by Cre, thereby enabling conditional reactivation of CFTR expression from the KI allele.

Generation of the CFTR
int1-eGFP(lsl) reporter ferret model.
Founder animals were generated by microinjecting ribonucleoprotein Cas9/sgRNA complexes and the donor DNA into ferret zygotes. We used mcDNA 19 as the donor of HITI 20 (Fig. 1B). This avoids the integration of an undesired plasmid backbone. Kits that were born from primipara pseudo-pregnant jills who received the microinjected embryos were genotyped by PCR using the primer sets L-Fw/int-rev and int-fw/R-Rev to amplify the left and right junction sequences of the insertion, respectively. One female kit, numbered AA2, yielded positive amplicons from both PCR reactions, indicating a potential founder. Upon Sanger sequencing analysis, no indels were found at the left junction. However, unexpected recombination occurred at the right junction. There was a 329 bp redundant sequence repeating part of the 5′ insert and a 251 bp deletion of the downstream intronic sequence.
To further investigate whether the AA2 kit could serve as a potential founder, we examined the integrity of the insert using Southern blotting with four phosphorus-32 (32P)-labeled probes, including two endogenous probes recognizing the upstream (left side, L) and downstream (right side, R) intronic sequences, as well as probes against the eGFP (G) and the 3′ UTR (U) sequence of CFTR within the insert (Fig. 1C). Genomic DNA was extracted from the blood of the AA2 kit at 2 months of age. For restriction enzyme digestions, we chose ApaL1, and the combination of ApaL1 and Kpn1. ApaL1 cleaves the CFTR locus to produce a 6.9 kb fragment encompassing the insertion site, but it does not cut within the insert. KpnI, on the contrary, cuts the insert once at the 3′ end of the UTR sequence. Southern blot analyses of DNA double-digested with ApaL1 and KpnI using the L and R probes revealed expected patterns, showing the 6.1 and 4.7 kb bands from the left and right junctions. However, a >25 kb band, rather than the expected 10.9 kb, was visualized from the Southern blot of the ApaL1-digested DNA. Based on the further analyses of the blots using the internal probes targeting the eGFP and the 3′ UTR, as well as the PCR reactions, which amplified the junction sequence from tail-to-head tandem repeated inserts and confirmed the absence of head-to-head or tail-to-tail repeats, we concluded that the AA2 ferret was inserted with the eGFP reporter cassette in a pattern of at least five head-to-tail copies in tandem (Fig. 1D).
The eGFP reporter functions as expected in airway epithelial cells from the AA2 ferret
We confirmed the targeted integration of the eGFP reporter at the desired site in intron 1. However, concern arose as it demonstrated a pattern of at least five head-to-tail copies in tandem. Thus, before using the AA2 ferret as the founder animal for line expansion, we characterized the functions of the insertion cassette. Using bronchoscope sampling, we obtained a tracheal biopsy from the AA2 ferret for primary airway epithelial cell cultures, which we used to examine eGFP reporter expression in CFTR-expressing cells and Cre-mediated lsl cassette removal from the KI allele. Since ionocytes express the highest level of CFTR in airway epithelium, we used immunofluorescence analysis (IFA) to assess ATP6VG3, 15 an ionocyte marker, and eGFP. As expected, we observed the localization of eGFP in ionocytes (Fig. 2A). The reporter in the KI allele is driven by the CFTR promoter, so transcription of eGFP mRNA was anticipated to be at a similar level as that of CFTR. To confirm this, we obtained total RNA from the ALI cultures. After cDNA conversion, the eGFP and CFTR transcripts were quantitated by TaqMan PCR using specific primer/probe sets, and data were normalized to the GAPDH transcript. As expected, transcript levels of eGFP and CFTR in these epithelium cultures were roughly equal, with a ratio of 1:1 (0.97 ± 0.01) (Fig. 2B).
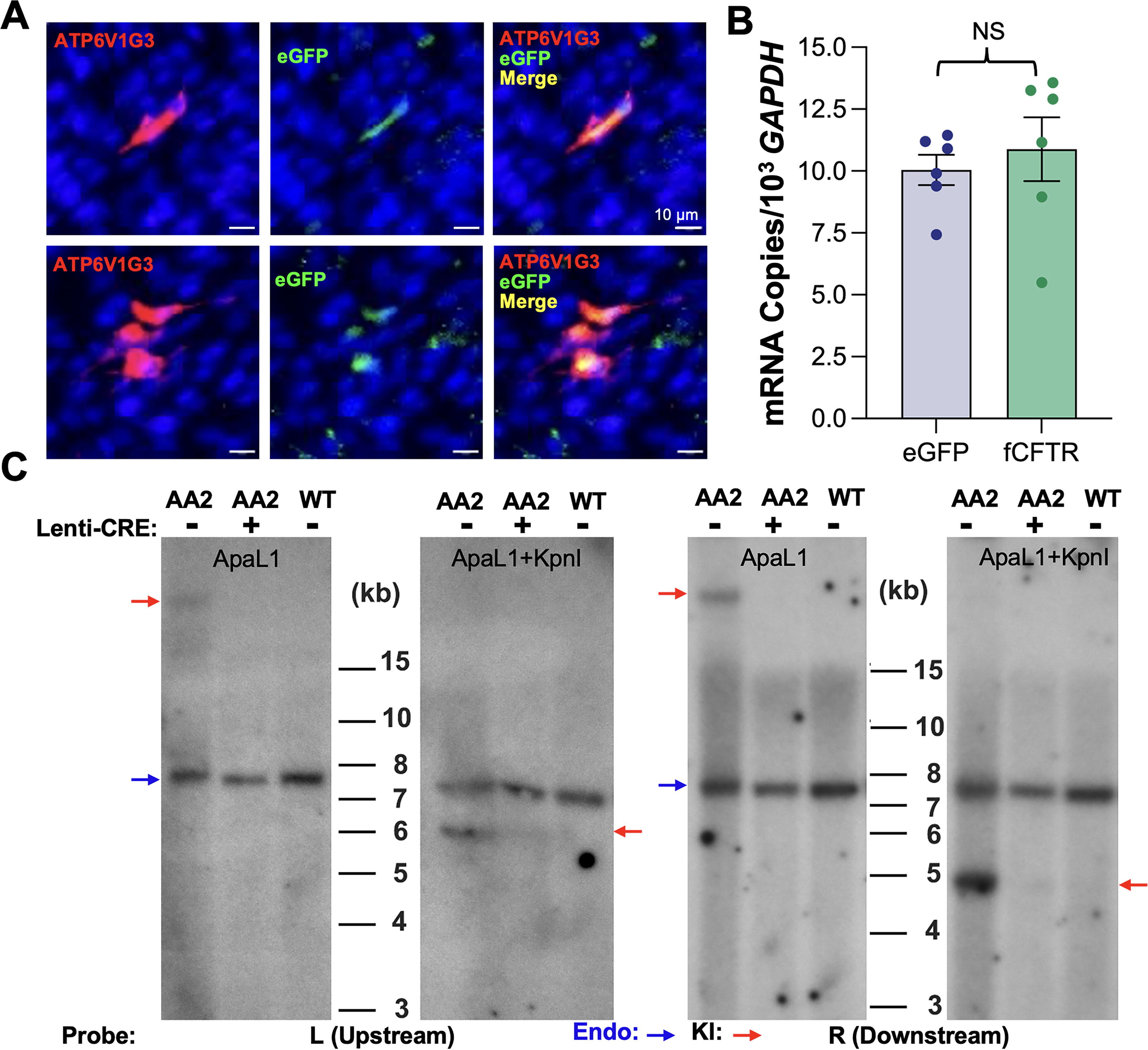
Functional characterization of the CFTR
int1-eGFP(lsl) founder.
Next, we transduced the primary airway basal cells with the vesicular stomatitis virus G protein (VSV-G)-pseudotyped lentiviral vector, HIVUbCpuroPGKCre, and treated cells with puromycin. Genomic DNA was extracted from puromycin-resistant cells and the control untreated cells for Southern blot analyses. Genomic DNA from a wild-type (WT) ferret was used as a control. Results confirmed that Cre expression effectively excised the lsl cassettes from the KI allele, although they were in the head-to-tail array of five tandem copies (Fig. 2C).
Cre exposure restored CFTR function in polarized epithelial cultures derived from CFTRint1-eGFP(lsl)/G551D CF ferrets
As the basic functions of the eGFP reporter were confirmed as anticipated, we bred the AA2 founder to WT ferret hobs. From two litters, six F1 kits (two males and four females) with the CFTR int1-eGFP(lsl)/wt genotype were identified by junctional PCR and Southern blot analyses, confirming a stable germline inheritance of the mutation. Next, we bred one of the F1 jills, to a heterozygous CFTR G551D/wt hob. We anticipated generating offspring with CF from this breeding and thus administered VX-770 to the pregnant jill during the gestation period to prevent the disease from developing. After birth, we identified three kits with the CFTR int1-eGFP(lsl)/G551D genotype. These were reared on VX-770 until they were weaned at 2 months of age. One of these ferrets was sacrificed due to health concerns resulting from pancreatic disease 22 days after VX-770 withdrawal. The other two ferrets were sacrificed at 3 months of age for experiments.
Primary airway epithelial cells isolated from the trachea and nasal respiratory epithelium of CFTR int1-eGFP(lsl)/G551D ferrets were cultured in PneumaCult Ex-plus medium to enrich the airway basal cells. A portion of the airway basal cells from both tissues were transduced with HIVUbCpuroPGKCre and selected with puromycin, respectively. Puromycin-resistant cells and control untreated basal cells were seeded onto Transwell inserts and differentiated at an ALI. We assessed transepithelial Cl- transport in mature polarized nasal and tracheal epithelium ALI cultures by measuring changes in transepithelial short circuit current (Isc) upon the addition of ion channel blockers or agonists using an epithelial voltage clamp and a self-contained Ussing chamber system, as previously described by our laboratory. 13,15 Isc changes were measured over the experiment period, which involved the sequential addition of amiloride, an epithelial sodium channel (ENaC) blocker, DIDS, a non-CFTR anion channel blocker, the forskolin and IBMX, which are cAMP agonists, and GlyH101, a CFTR-inhibitor (Fig. 3A). Forskolin and IBMX activated Cl- transepithelial transport, indicated as ΔIscF&I, and the induced CFTR-specific current was blocked by GlyH101, indicated as ΔIscGlyH101. Quantitative data from these measurements showed that the function of the CFTR-specific Cl− channel was detected in the tracheal and nasal ALI cultures derived from the Cre-treated airway basal cells (Fig. 3B). However, it was not detectable in the nontransduced CF ALI cultures. This indicates that the eGFP-KI allele regained function (i.e., CFTR expression was restored) by Cre-mediated excision of the reporter cassette in intron 1 through the recombination of the LoxP sites.
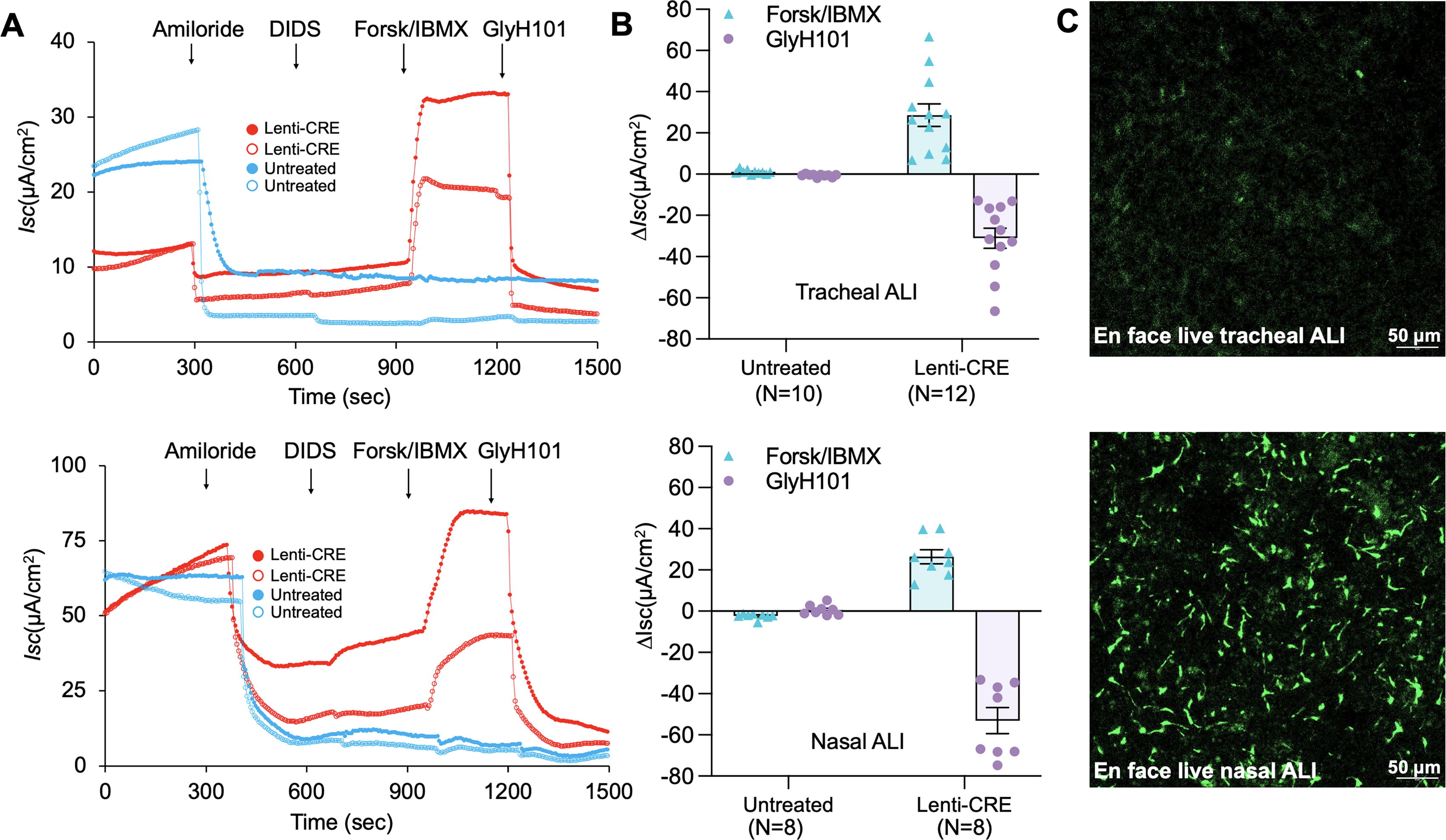
Cre recombinase restores the function of the eGFP reporter KI allele in polarized epithelial cultures derived from CFTR
int1-eGFP(lsl)/G551D ferrets.
To verify the expression of the GFP cassette in the tracheal and nasal cultures, we visualized eGFP expression under a confocal microscope. As expected, no eGFP-expressing cells were seen in ALI cultures derived from Cre-treated tracheal and nasal basal cells, whereas eGFP expression was observed in cultures from untreated cells. However, there were significantly fewer eGFP-expressing cells in the tracheal ALI cultures compared with the nasal ALI cultures, as shown in the representative en face images of the live cultures (Fig. 3C). Based on single cell RNA sequencing data, pulmonary ionocytes express the highest level of CFTR among airway epithelial cell types. 23,24 Given that eGFP expression was mostly associated with the ionocyte marker in tracheal ALI cultures (Fig. 2A), and the transcripts of eGFP and CFTR from the CFTR promoter were roughly at the same level (Fig. 2B), the greater numbers of eGFP-expressing cells in nasal cultures suggested that there was a higher abundance of ionocytes than in tracheal cultures. Thus, a higher level of CFTR expression is expected in nasal ALI cultures, compared with that in tracheal cultures. Nevertheless, although we anticipated greater CFTR expression in Cre-treated nasal ALI cultures, both types of Cre-treated cultures demonstrated similar levels of CFTR-specific transepithelial Cl- transport. It is unclear whether the ion channel activities of CFTR in nasal ALI are potentially less potent in conducting Cl- transport than those in the tracheal epithelium, which warrants further investigation.
eGFP reporter expression in polarized nasal epithelial cultures
Since we observed more eGFP-expressing cells in nasal ALI cultures, we wanted to investigate which cell type specifically expresses eGFP. Therefore, we used IFA to profile eGFP expression in different epithelial cells. First, nasal ALI cultures were costained with antibodies against eGFP, CFTR, and BSND an ionocyte marker 15 to confirm the coexpression of CFTR and eGFP in ionocytes. As expected, transduction with HIVUbCpuroPGKCre eliminated eGFP expression. In ALI cultures derived from untreated cells, most eGFP-expressing cells were BSND-positive ionocytes, where CFTR expression is positive (Fig. 4A). Notably, most BSND-positive ionocytes were associated with CFTR in both the Cre-treated and nontreated cultures, characterized by an “apical cap” area enriched with CFTR protein expression (Fig. 4A), similar to our previous observations. 15 Next, we probed the nasal ALI cultures with antibodies against other cell-type-specific markers: anti-Muc5B (a marker for goblet cells), anti-FoxJ1 (a nuclear marker for ciliated cells), and anti-acetyl-α-tubulin (AcTub, a surface marker for ciliated cells). 15 We performed IFA using high-resolution confocal microscopy, which enabled us to differentiate between cells expressing eGFP at high and low levels. In contrast with ionocytes, which were colocalized with high levels of eGFP, Muc5B-stained goblet cells were associated with low levels of eGFP, whereas no detectable eGFP was found in ciliated cells (Fig. 4B). ATP6V1G3 staining demonstrated colocalization with CFTR, and eGFP confirmed that ionocytes express high levels of both CFTR and eGFP (Fig. 4C and D).
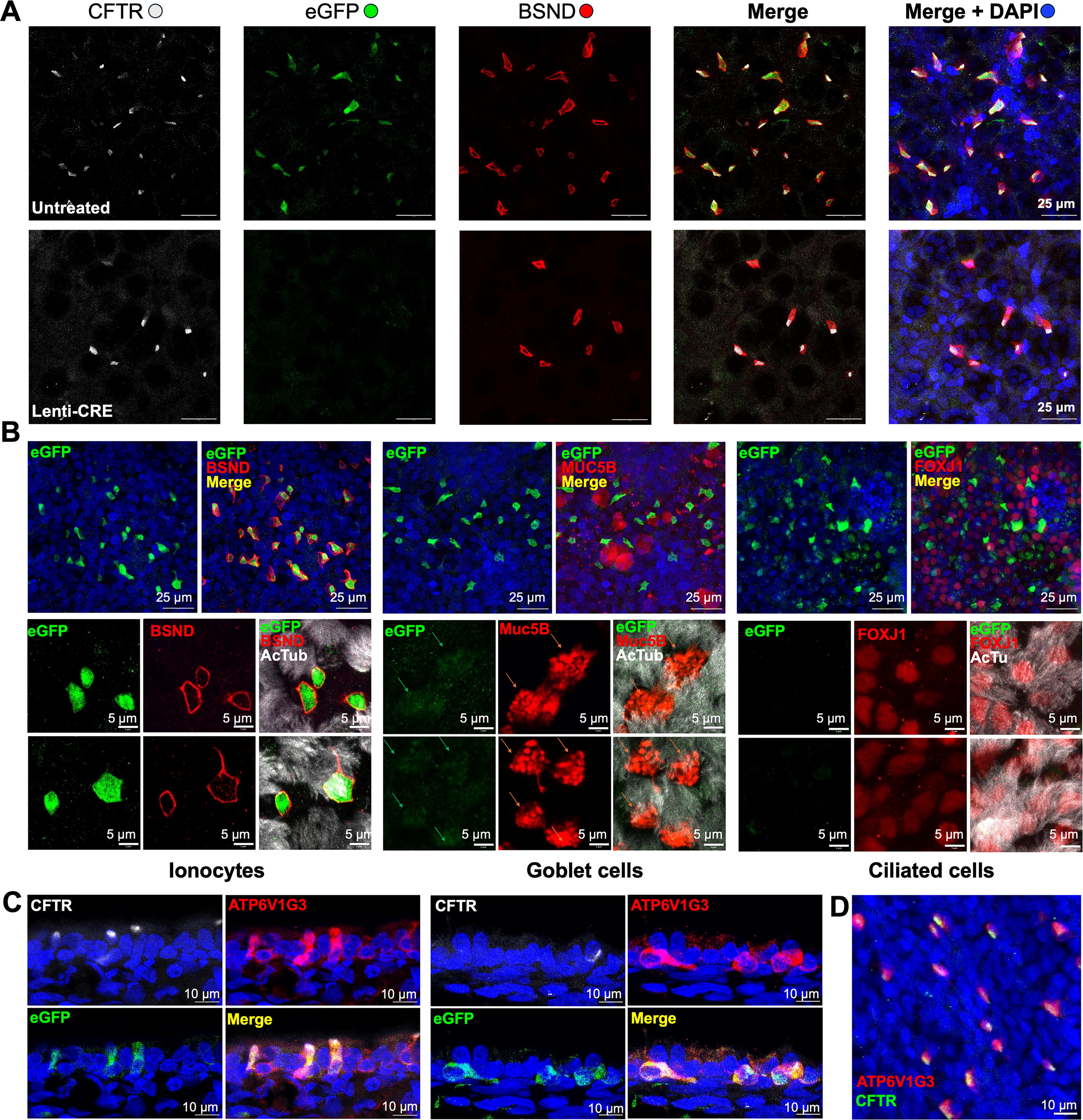
Cell-type-specific eGFP reporter expression in polarized nasal epithelial cultures from CFTR
int1-eGFP(lsl)/G551D ferrets. Nasal ALI cultures derived from CFTR
int1-eGFP(lsl)/G551D ferrets that were transduced with HIVUbCpuroPGKCre and selected with Puro, or not transduced (untreated). The ALI cultures were subsequently costained with antibodies against CFTR and BSND (barttin CLCNK type accessory subunit, an ionocyte marker), as well as selected epithelial cell markers.
eGFP reporter expression in selective ferret tissues/organs
Recent research has revealed a gradient of ionocytes from the proximal to distal regions of the airways. In the nasal mucosa, ionocytes can reach up to 3% of the total cell population, whereas they are rare in the bronchi. 25 Using whole mount tissue staining, we assessed eGFP-expressing cells in the nasal respiratory epithelium, trachea, and other CFTR-rich organs such as the pancreas. Immunostaining for the ionocyte marker, BSND, and CFTR confirmed highly abundant ionocyte numbers in the nasal respiratory epithelium (Fig. 5A). Notably, we found CFTR+BSND+ ionocytes expressed eGFP (Fig. 5A). We next assessed eGFP expression in the trachea and found the ionocyte marker ATP6V1G3 (H+ transporter) 15 was colocalized with eGFP (Fig. 5B). The pancreas is particularly affected by CF, with CFTR mRNA being expressed in the exocrine cells of the ferret pancreas, but not in the INS-expressing endocrine cells. 26 By IFA, we observed the colocalization of CFTR and eGFP in the exocrine cells (Fig. 6A) and pancreatic ducts (Fig. 6B), which is consistent with our previous studies demonstrating CFTR mRNA in the pancreas is exclusively in exocrine cells and no evidence of CFTR expression in INS-expressing endocrine cells. 26 Notably, CFTR and eGFP demonstrate significant enrichment within SOX9-positive centroacinar cells in the pancreas (Fig. 6C–E), but not in INS-expressing beta cells (Fig. 6A and F).
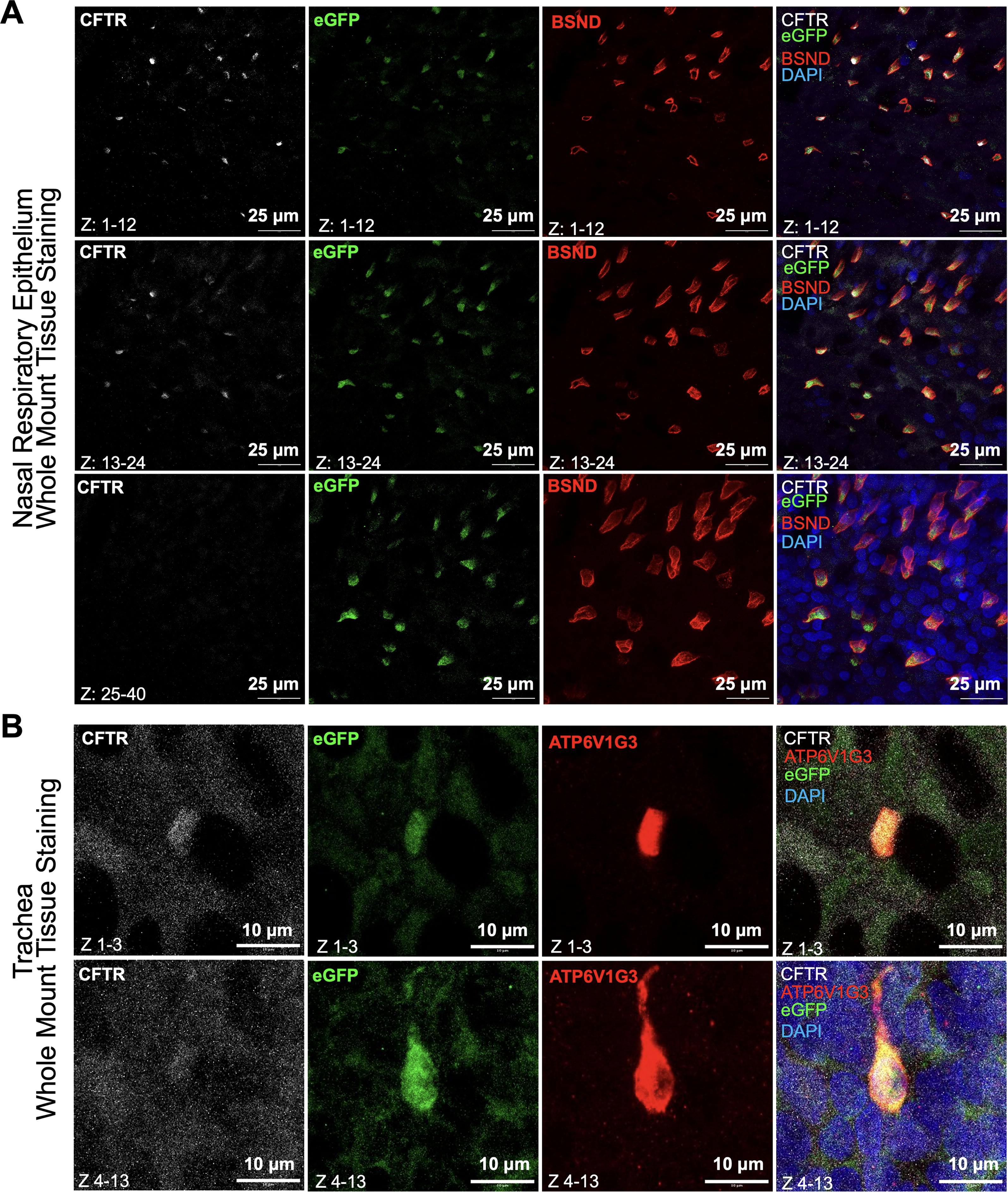
Colocalization of eGFP and CFTR in ionocytes of airway epithelium.

Colocalization of eGFP and CFTR in centroacinar and ductal epithelial cells of the pancreas.
DISCUSSION
CF is a recessive genetic disorder, and the use of gene therapy to add back a normal copy of the CFTR gene to deficient cells holds great potential for ultimately curing the disease. However, this approach is challenging due to the heterogeneity of CFTR expression in multiple cell types in the lungs. To better understand where and when CFTR is expressed in cells in the airways, we developed the CFTRint1-eGFP(lsl) reporter ferret, in which we can control CFTR expression in a cell-type-specific manner throughout development.
The draft ferret genome sequence was published, and the genome assembly was annotated using the ESEMBL gene annotation system. 27 While knowledge of ferret CFTR expression regulation and its structural and regulatory elements is limited, extensive studies on human CFTR regulation and DNA modifications provide valuable insights. 28 –31 Notably, investigations of human CFTR intron 1 identified that regulator elements, including a prominent DNase I-hypersensitive site (DHS) at +10 kb and weak DHSs at +12 and +14 kb (to exon 1), exhibit enhancer activity in a tissue-specific manner. 32 Comparable studies have not yet been conducted for the ferret genome. To minimize potential impacts on disrupting regulatory elements, we selected a target site near the 3′ end of intron 1, ∼1 kb from exon 2, to avoid such DNA hypersensitivity sites. BLAST® (basic local alignment search tool) analysis revealed a 1.5 kb sequence of the 3′ end of ferret intron 1, including a 500 bp sequence surrounding the sgRNA3 recognized site, and showed no significant homology with the 27,245 bp sequence of mouse CFTR intron 1. To further validate our selection, we tested three sgRNA targeting the region ∼1 kb upstream of exon 2, using in vivo cleavage assays via zygote microinjection of CRISPR complex. Offspring from the transplanted embryos (treated) exhibited normal phenotype, like the untreated controls, suggesting that non-homologous DNA end joining (NHEJ)-mutagenesis at these sites (when occurring at two alleles) did not significantly disrupt the CFTR gene function. Based on these results, the best performing sgRNA (gRNA3) was chosen for this study.
This new model will facilitate future studies to uncover the specific roles of CFTR in regulating airway clearance and innate immune responses within the context of disease. Single-cell RNA sequencing (scRNAseq) suggests that pulmonary ionocytes express the highest level of CFTR in the large and small airways, 23,24 whereas secretory cells and ciliated cells exhibit low or negligible levels of CFTR expression. 33,34 Consistent with these findings, our analyses of eGFP reporter expression in polarized airway epithelial cultures in vitro and the whole mount tissues of trachea and lung from the CFTR int1-eGFP(lsl) reporter ferret demonstrated that intrinsic CFTR promoter-driven eGFP signals were highly enriched in pulmonary ionocytes. We also observed lower levels of eGFP expression in a small subset of MUC5B-positive goblet cells, but little to no signal was found in ciliated cells. Goblet and ciliated cells are just a subset of the cell types present in the airways. Thus, further studies identifying the extent of coexpression of eGFP and CFTR in vivo in different tissues/organs will help shed light on cellular phenotype heterogeneity within CFTR expressing cells.
Our recent study using ferret models in which CFTR expression can be deleted specifically in ionocytes suggests that rare pulmonary ionocytes perform critical CFTR-dependent functions in the proximal airway and disrupting this function results in hallmark defects in surface airway fluid regulation as observed in individuals with CF. 15 This suggests that restoring CFTR expression, specifically in ionocytes, might improve CF defects in mucociliary clearance of the proximal airways. However, from a gene therapy standpoint, targeting rare populations like ionocytes, which comprise only 0.5–1.5% of epithelial cells along the large conducting airways, presents a significant challenge. Since ionocytes are not the sole CFTR-expressing airway epithelial cell type, the necessity of CFTR transfer to ionocytes for functional complementation remains unclear. In contrast, ciliated cells have long been considered a potential target for CF gene therapy, as they are abundant on the airway surface and accessible to the vectors delivered through the apical lumen. Moreover, CFTR transfer to ciliated cells has demonstrated the ability to correct Cl− transport deficiency in vitro in polarized human airway epithelial ALI cultures derived from CF patients. 35 However, now we know that ciliated cells express little to no CFTR mRNA. Thus, it remains uncertain whether ectopic CFTR expression in this cell type can sufficiently compensate for lung function in patients with CF. Although gene therapy is a promising potential approach to treat CF, it remains unclear which cell types should be targeted to restore CFTR expression. This is a significant knowledge gap that must be overcome before this approach can be successfully implemented. This necessitates a deeper understanding of the pathophysiology and pathogenesis of CF lung disease and how CFTR functions at the cellular level to control airway clearance and innate immunity.
Our novel CFTR int1-eGFP(lsl) reporter ferret model, in which CFTR expression is disrupted but can be conditionally reactivated, can be used in conjunction with existing FOXI1-CreERT2 driver ferrets and other cell-type-specific Cre driver lines under development, such as the MUC5B-CreERT2 and SFPTC-CreERT2 ferrets, to assess CFTR function in ionocytes, goblet cells, and alveolar type 2 (AT2) cells. These studies allow for the selective reactivation of CFTR expression in specific epithelial cell types in the context of CF and facilitate lineage tracing to determine the fate of CFTR-expressing cells. They will provide a unique and invaluable resource in which to advance our understanding of the pathophysiology of CF and to identify novel targets for gene therapy. Additionally, the CFTR int1-eGFP(lsl)/G551D CF model also provides a platform for testing gene therapies using exogenous Cre gene transfer to prevent the progression of CF lung disease (by delivering Cre prior to VX-770 withdrawal) or to reverse lung disease (by delivering Cre in the context of CF after VX-770 withdrawal). By breeding CFTR lsl-eGFP/G551D ferrets with the ROSAmTmG ferrets, 36 the incorporation of the fluorescence-convertible reporter that is also Cre-responsive enables us to genetically label the CFTR-reactivated cells followed Cre gene transfer.
With the advancement of CRISPR-based genome editing techniques, gene therapy is progressing from functional complementation via gene addition to the correction of defects in disease-causing genes at the genomic level. 37 Several CRISPR-based therapies are currently in clinical trials. 38,39 Gene editing offers the potential to permanently correct defective genes without altering their natural expression patterns. For CF gene therapy, precise repair of the defects in the endogenous CFTR gene addresses the limitation of the traditional CFTR replacement, which lacks regulation. Although gene editing in terminally differentiated epithelial cells can restore CFTR expression at endogenous level for the lifespan of these cells, permanent correction can be achievable by targeting lung progenitor cells, such as basal cells, club cells, and AT2 cells. 40 These cell types can self-renew and differentiate into various cell types that express CFTR at physiological levels, making it possible to maintain pulmonary homeostasis and innate immunity.
Collectively, the CFTR lsl-eGFP/G551D ferret can serve as a valuable model system to advance research on CFTR lung biology, CF pathogenesis, and CF gene therapy. It provides a powerful tool for assessing the delivery efficiency of gene editing vectors and therapeutic efficacy of CFTR gene editing approaches.
Footnotes
AUTHORS’ CONTRIBUTIONS
F.Y.: Conceived, design and conducting experiments, data collection, analysis assembly, and interpretation. X.S.: Ferret model generation. Z.Y.: Transgenic construct design. S.Y.P. and Z.F.: Ferret model molecular characterization. Y.Y.: Ferret model breeding. F.Y. and S.Y.P.: Polarized epithelial culture, collection, analysis, and interpretation of electrophysiology data. F.Y., S.Y.P., Y.T., M.E., A.E.T., and J.D.K.: Collection, analysis, and interpretation of data of cell culture staining and tissue staining. F.Y., J.F.E., and Z.Y.: Writing the article with input from all authors. J.F.E. and Z.Y.: Conceptualization, funding acquisition, supervising research, and final approval of the article.
AUTHOR DISCLOSURE
The authors declare no conflicts of interest.
FUNDING INFORMATION
This work was funded by the following grants from the National Institutes of Health (R01 HL174593, R21 AI182645 to Z.Y.; P30 DK054759, RC2 DK124207, P01 HL152960, and R01 HL165404 and Federal Contract 75N92024C00008 to J.F.E.), the Cystic Fibrosis Foundation (YAN23G0 to Z.Y.; ENGELH21XX0 to J.F.E.; and 004590G222 to X.S.), and the Cystic Fibrosis Research Institute (Grant#1267026 to F.Y.).