
Abstract
The development of potency assays for Advanced Therapy Medicinal Products (ATMPs) presents significant challenges due to the variability of starting materials and the complex mechanisms of action involved. This article aims to address the following key question: How can we design robust and reliable potency assays for ATMPs that accommodate product-specific challenges and align with evolving regulatory standards? To answer this, we employed a mixed-methods approach, synthesizing data from scientific literature, industry reports, and regulatory guidelines to identify current limitations and innovative solutions for potency assay development. Our methodology integrates a systematic review of academic publications (2018–2024) to capture recent advancements in biotechnology and their applicability to potency testing. We complemented this with an analysis of industry perspectives, drawn from webinars and white papers, as well as a detailed comparison of global regulatory frameworks, including the FDA’s new guidance on potency assurance for Cellular and Gene Therapy Products (CGTs/ATMPs). Additionally, we developed a comprehensive database to analyze potency assays used in approved, rejected, and withdrawn CGT/ATMP products, focusing on technical and regulatory challenges. Based on this multilevel analysis, we propose a product-specific framework for designing, developing, and validating potency assays for different ATMP categories, taking into account their unique technical and regulatory constraints. We also highlight emerging technologies, such as droplet digital polymerase chain reaction and reporter gene assays, as innovative tools for improving the precision and reliability of potency testing. Our findings underscore the need for flexible, risk-based strategies in potency assay development that evolve throughout product development and clinical trial phases. Future recommendations emphasize assay standardization, the definition of acceptable variability, and stronger correlations between in vitro potency data and clinical outcomes.
Keywords
INTRODUCTION
Cell and Gene therapies (CGT, as referred to by the Food and Drug Administration [FDA]) 1 or Advanced Therapy Medicinal Products (ATMP, as referred to by the European Medicines Agency [EMA]) 2 are innovative therapies that mainly target orphan diseases and high unmet medical needs. They include gene therapy medicinal products (GTMP), cell therapy medicinal products (CTMP), tissue-engineered therapies (TEP), and combined ATMP. Combined ATMPs as defined by the EMA are a class of treatments that may contain one or more medical devices as an integral part of the medicine to address complex health conditions. Key therapeutic areas addressed by ATMPs include genetic disorders, hematological malignancies, and other cancers or long-term diseases, monogenic diseases, and cartilage diseases. 3 Since legislation opened the way in 2007, only 27 ATMPs have been approved in Europe by the EMA, and 38 CGTs approved by the U.S. FDA, some of them have already been withdrawn. In addition, mRNA therapies represent a groundbreaking approach to treating a variety of diseases, including infectious diseases, cancer, and genetic disorders.4,5 While demonstrating significant potential, these therapies navigate a complex regulatory landscape. While both the European Union and United States can classify mRNA-based therapeutics as gene therapies, the specific definitions and implications of this classification vary significantly between regions, further emphasizing the need for harmonized regulatory frameworks in this rapidly evolving field. 6
The complex nature of CGT/ATMP poses significant Chemistry, Manufacturing, and Control (CMC) challenges to establish well-defined and standardized analytical methods for products characterization, ensuring accurate assessment of product quality, identity, purity, and potency. 7 CMC challenges for ATMPs differ significantly from traditional drugs due to their unique characteristics. ATMPs, which often involve living cells, gene therapy vectors, or personalized manufacturing, require solutions for maintaining cell viability, ensuring gene vector quality, controlling contamination, and navigating evolving regulatory frameworks. Additionally, scaling up and post-market monitoring are additional challenges due to the complexity of maintaining consistency in larger-scale manufacturing processes for living cells or gene vectors, and the need for long-term monitoring of safety and efficacy in patients over time. Additionally, post-market monitoring requires ongoing assessment of product stability, efficacy, and potential adverse events in a real-world setting. These complexities demand innovative approaches and strict quality control measures, necessitating collaboration between industry, regulators, and research institutions to ensure the safe and effective production of ATMPs.
As part of the CMC process, according to international guidance, potency is the quantitative measure of biological activity, which is linked to the relevant biological properties of the product. 8 It determines the product’s ability to produce the desired therapeutic effect or elicit an appropriate immune response. Potency assays are crucial in the development of therapeutic products as they provide objective measurements of product efficacy, ensuring consistent quality and regulatory compliance. They help in product characterization, lot release testing, batch-to-batch consistency, comparability studies, and quality control, playing a vital role in ensuring the potency, safety, and effectiveness of the final product. 9 A combination of multiple methods may be needed to adequately define potency in functional assays during product development programs. Certain assays may be needed to control quality amid procedural changes, whereas others are more suitable for product characterization, comparability, and release for clinical use. 10
Without reliable potency assays, it would be impossible to assess the safety and efficacy of CGTs/ATMPs, and the development of these life-saving therapies would be hindered. If the potency assays for a CGT/ATMP product are not adequately validated or if they do not provide a clear measure of the product’s strength or activity, the EMA and FDA may delay approval of the product.11–14
The EMA and FDA’s guidelines on potency assays for CGTs/ATMPs state indeed that the potency of an ATMP is a critical quality attribute (CQA) and must be demonstrated to be precise, consistent, and reproducible throughout the manufacturing process. 15 The International Council for Harmonisation (ICH) Q14 guideline, officially titled Analytical Procedure Development, was developed by the ICH 16 to provide a framework for the systematic development of analytical procedures. It focuses on ensuring that methods used to assess product quality (such as potency assays) are scientifically justified, robust, and optimized for regulatory approval. ICH Q14 emphasizes a lifecycle approach to analytical methods, encouraging the use of risk-based principles, such as Design of Experiments (DoE), to enhance the reliability of assays. Its goal is to improve the reproducibility and flexibility of analytical procedures across product development stages, ensuring that quality is maintained from early phases to post-marketing. 17
For ATMPs, implementing ICH Q14 poses challenges due to the inherent complexity and variability of these therapies, requiring tailored solutions for potency assay development.
In addition to these guidelines, the EMA and FDA also have specific requirements for the potency assays of different types of ATMPs. For example, the potency assay for a gene therapy product will be different from the potency assay for a cell therapy product.8,11–13,18–20
The newly released guidance from the FDA introduces a “potency assurance strategy” as a comprehensive approach to help ensure that every lot will have the potency necessary to achieve the intended therapeutic effect, tailored for CGT products. 21 Unlike the previous 2011 guidance, which primarily focused on potency tests, this updated and broader approach underscores the necessity of a comprehensive strategy throughout the entire lifecycle of product development. It aims to accommodate the diverse nature of CGT products and encourages a flexible and adaptable approach. Key features of this strategy include developing a Quality Target Product Profile, identifying potency-related CQAs, and implementing a formal risk assessment process. The strategy should be multifaceted, addressing risks from manufacturing through post-release stages, with an emphasis on reducing risks to potency-related CQAs.
Furthermore, the document addresses communication with the FDA, challenges in controlling potency, identification of potency-related CQAs, and offers specific recommendations for different product classes. It also provides detailed recommendations on assessing potency, qualifying, and validating assays, and managing changes throughout the product lifecycle.
General principles of potency assay development for ATMP have been published in prior guidelines and literature.11–13,18,21,22 This article aims to address the complexities of potency assay development for ATMPs by focusing on the necessity of tailored approaches for different product types and by synthesizing knowledge from diverse sources, including scientific literature, industry reports, and regulatory guidelines. Through a comprehensive analysis of approved, rejected, and withdrawn ATMPs, we propose a product-specific framework for designing, developing, and validating potency assays. This article offers practical insights and recommendations, enriched with real-world examples, to guide industry stakeholders in ensuring consistent quality and regulatory compliance throughout the lifecycle of different type of ATMPs.
METHODS
First, we conducted a systematic academic literature review on potency assays for CGT/ATMP development, focusing on scientific and regulatory challenges. This review encompassed papers published between 2018 and November 2024, aiming to identify scientific innovations that may offer solutions to potency assay challenges. To conduct this systematic review, a comprehensive literature search was performed across multiple databases, including PubMed and Google Scholar, to identify peer-reviewed articles, reviews, and industry reports related to potency assay development for ATMPs. The search covered publications from January 2018 to December 2024 and employed combinations of relevant keywords such as “potency assay,” “advanced therapy medicinal products,” “gene therapy,” “cell therapy,” “biological products,” and “regulatory challenges.” Boolean operators (e.g., “potency assay” AND “gene therapy”) were used to refine search results. Articles published in French and English were included. Inclusion criteria encompassed studies explicitly addressing methodologies, challenges, or advancements in potency assays for ATMPs, including clinical studies, case reports, and regulatory guidelines. Studies unrelated to ATMPs, lacking clear methodologies, or focusing solely on chemical drug potency were excluded. A total of 152 primary studies were identified, with 47 meeting the inclusion criteria after screening titles, abstracts, and full texts. Data were extracted based on study type, therapeutic focus (gene therapy, cell therapy, or combined ATMPs), assay techniques, and references to regulatory guidelines. The review revealed a significant emphasis on functional assays (47%), biomarker-based approaches (22%), and advanced analytical methods (31%). Approximately 63% of the studies referenced specific regulatory perspectives from agencies such as the FDA and EMA. This methodological framework facilitated a comprehensive analysis of key trends, challenges, and recommendations for potency assay development in ATMPs.
Second, we systematically analyzed industry reports from cell and gene therapy webinars and white papers published by providers and pharmaceutical industries during the same period. This analysis aimed to capture industry perspectives and advancements in potency assay development. To complete this view, we established a comprehensive database of potency assays for approved, rejected, and withdrawn CGT/ATMP products, utilizing regulatory and scientific data. This database focuses on technical and regulatory issues, including the main reasons for product withdrawals, sourced from the European Public Assessment Report available on the EMA website.
By following this multipronged approach, we were able to gather a comprehensive understanding of the current landscape of potency assays in CGT/ATMP development, including the scientific and regulatory challenges, industry perspectives, and the role of these assays in product approval and withdrawal decisions. This information allowed us to build and propose a framework to address these challenges and ensure the robust assessment of potency in CGT/ATMP therapies taking into account product type and throughout the different stages of product development.
RESULTS
Common challenges in CGTs/ATMPs potency assay development
The implementation of ICH Q14 principles for potency assays in ATMPs faces several challenges. 16 The inherent variability of starting materials, including autologous and allogeneic donor variability and cell line heterogeneity, poses difficulties in establishing consistent potency assays for CGTs/ATMPs Moreover, the presence of error-prone replicating viruses further contributes to the variability. Limited batch size and material availability for testing, especially in single-dose therapies using autologous cells, create constraints in developing robust potency assays. Additionally, the limited stability of cellular products, such as viability issues, adds to the complexity of assessing their potency during storage and transportation. 23 Regulatory bodies recognize that cellular starting material can be highly variable due to process and donor variability. As a result, it is critical that starting material is collected, stored, shipped, and tested with a highly reproducible process.
Using a supplier who is incapable of manufacturing or supplying GTMP (Gene Therapy Medicinal Products)/Good Manufacturing Practice (GMP)-compliant cellular starting materials will cause a significant delay in the speed at which a therapy reaches the market.
Furthermore, a significant challenge is the lack of appropriate reference standards for autologous cellular material and novel gene therapy vectors, making it challenging to establish standardized potency assays. CGTs/ATMPs often consist of multiple active ingredients, such as multiple cell lines or heterogeneous mixtures of peptide-pulsed tumor and immune-modulatory cells, along with the use of multiple vectors in combination 10 (Fig. 1).

Factors that influence cell and gene therapies product potency. GTMP, Gene Therapy Medicinal products; CTMP, Cell Therapy Medicinal Products.
Understanding the in vivo fate of the product, such as migration from the site of administration, cellular differentiation into the desired cell type, viral or cellular replication, uncoating, and transgene expression, further complicates potency assessment. 24 To overcome these challenges, innovative approaches and tailored analytical methodologies are required to develop reliable potency assays that accurately assess the therapeutic efficacy of CGTs/ATMPs. In Table 1, we discuss possible solutions for main potency assay challenges for CGTs/ATMPs.
Potency assay challenges for cell and gene therapies and their possible solutions

AAV, adeno-associated virus; ATMP, Advanced Therapy Medicinal Product; AUC, Analytical ultracentrifugation; CAD, charged aerosol detection; CGT, Cellular and Gene Therapy; icIEF, Imaged capillary isoelectric focusing; LC-HRMS, liquid chromatography-high resolution MS; PTM, Post-Translational Modification; UHPLC, ultrahigh-performance liquid chromatography.
Current landscape of potency assay
The complexities and challenges outlined in the development of potency assays for CGTs/ATMPs, such as variability in starting materials and the lack of appropriate reference standards, highlight the need for innovative solutions, and tailored analytical approaches. As these challenges are navigated, it becomes crucial to explore the current landscape of potency assays that have been validated for approved products by regulatory bodies like the EMA. Understanding the diversity and specificity of these validated assays, as shown in Table 2, provides valuable insights into how these challenges can be addressed effectively. Different types of potency assays can be used for CGTs/ATMPs. In vivo potency assays address animal death, survival, and tumor shrinkage. Ex vivo assays study dose administration, cellular response, and cytokine profiling. In vivo and ex vivo assays include the measurement of cell proliferation, growth arrest, cell death, motility, differentiation, and infectivity on another level alteration of intracellular pathways and the presence of active transgene products can be measured. Finally, expression of transgenes and, where relevant, alteration in transcriptome may be measured to provide additional insights into potency. 10
List of all potency assays used for advanced therapy medicinal product and validated by EMA
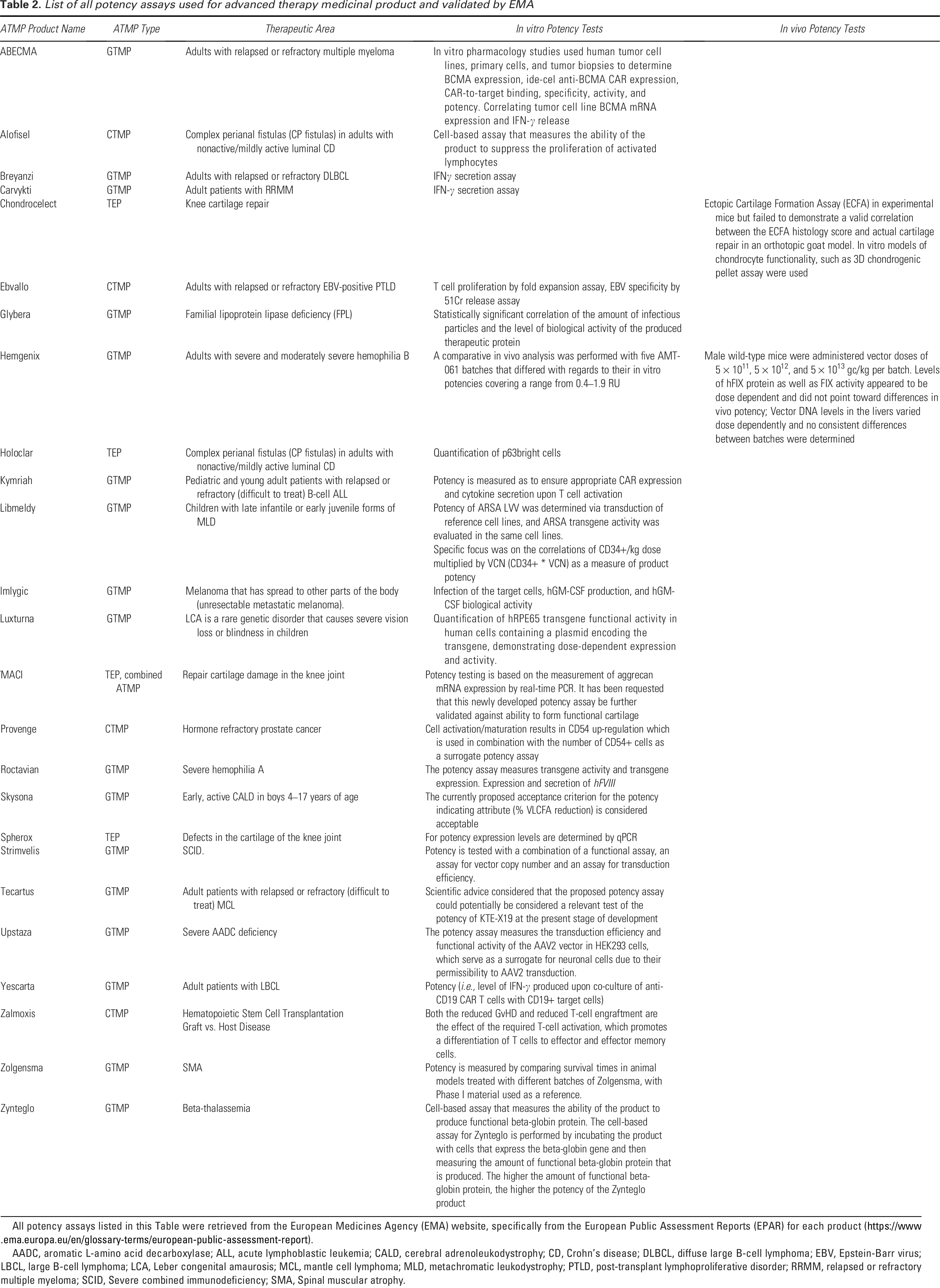
All potency assays listed in this Table were retrieved from the European Medicines Agency (EMA) website, specifically from the European Public Assessment Reports (EPAR) for each product (https://www.ema.europa.eu/en/glossary-terms/european-public-assessment-report).
AADC, aromatic L-amino acid decarboxylase; ALL, acute lymphoblastic leukemia; CALD, cerebral adrenoleukodystrophy; CD, Crohn’s disease; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; LBCL, large B-cell lymphoma; LCA, Leber congenital amaurosis; MCL, mantle cell lymphoma; MLD, metachromatic leukodystrophy; PTLD, post-transplant lymphoproliferative disorder; RRMM, relapsed or refractory multiple myeloma; SCID, Severe combined immunodeficiency; SMA, Spinal muscular atrophy.
The potency tests approved by EMA vary across products, reflecting the diversity of ATMPs and their specific mechanisms of action. Some tests involve sophisticated in vitro studies, while others use more straightforward measures like IFN-γ secretion or cell proliferation. Correlation between in vitro and in vivo potency is highlighted in some cases, as it is important to assess the relevance of in vitro assays to clinical outcomes. For example, in the case of Hemgenix, a GTMP used for the treatment of adults with severe and moderately severe hemophilia B, the potency test used was a comparative in vivo analysis using different batches with varying in vitro potencies.25,26 Despite up to almost 5-fold differences in in vitro potency among different batches, all batches produced a clear dose-related pharmacodynamic (PD) effect that was comparable within each dosing level in an in vivo setting. In this case, the in vitro potency differences did not significantly impact the actual PD effects observed in vivo. This finding suggests that, in this context, the in vitro potency assay used may not be a critical determinant of the therapeutic efficacy observed in the animal model.
Finally, this table shows that some products employ well-established assays, while others may require further validation or reevaluation based on post-approval data. 25
POTENCY ASSAYS FOR ADVANCED THERAPY MEDICINAL PRODUCTS: POTENCY ASSAY STRATEGIES AND BEST PRACTICES
General recommendations for potency assay development
The discussion on the current landscape of potency assays provides an essential context for understanding the practical application and regulatory expectations surrounding these tests in CGTs/ATMPs. This foundational knowledge sets the stage for exploring best practices and strategic recommendations for potency assay development. These recommendations aim to address the complexities inherent in cell and gene therapies, emphasizing the need for assays that not only measure relevant functional attributes but also comply with evolving GMP standards throughout the drug development process. By transitioning from the analysis of existing assays to the strategic approaches for developing robust assays, the text highlights the importance of both learning from established methodologies and innovating to meet the specific challenges posed by these advanced therapies. This progression underlines the critical role of tailored potency assays in ensuring therapeutic efficacy, regulatory compliance, and successful clinical outcomes as the field of CGTs/ATMPs continues to evolve. On one hand, we have seen that potency assays should account for and address inherent heterogeneity in these therapies. They should measure relevant functional attributes that ideally correlate with therapeutic efficacy. However, the complexity of cell and gene therapy can make quantifying potency tricky. On the other hand, GMP requirements are mandatory for licensure, and there is an increasing expectation for assuring product quality and consistency of manufacturing, product characterization, and compliance with current GMP (cGMPs: which focus more on the latest quality standards) as the drug development pipeline progresses. During phase 1 of development, the focus is largely on product safety issues. During phase 2, there are increased expectations for product characterization and compliance with cGMPs. During Phase 3, the focus is to ensure product quality and consistency of manufacturing and for the last stages full compliance is required for licensure.
The new FDA guidance 21 recommends an effective control strategy for materials and process parameters, recognizing the need for flexibility in certain products, where different strategic levers could be leveraged. This strategy acknowledges the diverse nature of CGT products and allows for flexibility in control approaches.
The advocated life cycle approach should start with early-stage strategies expected to be less comprehensive but still requiring a risk assessment. As development progresses, the strategy should evolve, incorporating insights from accumulated experience, clinical data, and stability information. Revisiting the strategy before licensure submission is crucial. The application of DoE in the development of analytical methods for ATMPs, such as AAV-mediated gene therapies, plays a critical role in optimizing assay performance and managing its lifecycle. DoE enables systematic and efficient exploration of multiple factors that influence assay outcomes. For example, it can address the interference of empty capsids in infectivity assays by optimizing conditions that minimize their impact on results. By identifying and controlling variables, DoE enhances robustness, accuracy, and reproducibility of potency assays. This approach aligns with regulatory expectations for method validation and lifecycle management, ensuring consistent therapeutic efficacy and safety. 27
Potency testing is crucial in determining the appropriate dose for proof-of-concept (PoC) studies in ATMPs. By quantitatively measuring the biological activity of a product, potency assays provide critical insights into the therapeutic efficacy at different dose levels. This data informs the selection of an optimal dose that maximizes efficacy while minimizing toxicity. Potency testing helps establish a dose-response relationship, guiding adjustments in dosing regimens for early-stage clinical trials and ensuring that the selected dose is both safe and effective.
For example, in AAV-mediated gene therapies, potency assays can help identify the minimal effective dose that achieves the desired gene expression levels without significant off-target effects. This approach aligns with regulatory guidelines, ensuring that the therapeutic dose used in PoC studies is scientifically justified and supports subsequent phases of clinical development.18,27
Tailored approaches for different product types, including cellular products, short shelf-life products, viral gene therapy vectors, vector-transduced cellular products, and tissue-engineered medical products, are presented. These approaches offer flexibility, with alternative methods considered acceptable. For cellular products, early identification of attributes linked to the intended therapeutic effect using nonclinical data is highlighted. Short-shelf-life products benefit from in-process testing for potency attributes, followed by post-release bioassays. Potency assays for Viral gene therapy vectors focus on transgene mRNA or protein, with attention to risks associated with vector particles and nucleic acids. Vector-transduced cellular products include potency assays for cellular and vector release. Tissue-engineered medical products collect comprehensive data on biological activity and use assays to address potential risks. Potency assays are also useful for assessing stability, device compatibility, and comparability after manufacturing changes, with criteria chosen through a quality risk management approach.
In the following, we discuss recommendations for potency assay for each type of CGT and their product development stage, along with innovative solutions for their development challenges.
GENE THERAPY: POTENCY ASSAY CHALLENGES AND RECOMMENDATIONS
In both the European Union and United States, protein expression alone is not sufficient for marketing authorization of biologics or gene therapies. Biological activity must be demonstrated to confirm that the product is functional and achieves its intended therapeutic effect. Regulatory guidelines, such as the EMA’s Guideline on the Quality, Nonclinical, and Clinical Aspects of Gene Therapy Medicinal Products (2018) and the FDA’s Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products (2011), emphasize that potency assays must reflect the product’s mechanism of action (MoA) and therapeutic efficacy. For gene replacement therapies, such as those targeting structural proteins like dystrophin in Duchenne Muscular Dystrophy (DMD), regulatory agencies require evidence of both protein expression (e.g., via immunohistochemistry or Western blot) and functional activity (e.g., restoration of muscle integrity, improvement in muscle function, or integration into the dystrophin-associated protein complex). In the case of DMD, clinical trials for micro-dystrophin therapies (e.g., Sarepta Therapeutics’ SRP-9001) 28 have demonstrated the importance of combining quantitative protein expression data with functional endpoints, such as improved muscle strength or histopathological evidence of muscle fiber restoration. These requirements ensure that the therapy not only produces the target protein but also achieves its intended therapeutic effect. Gene therapies have two steps that require potency quantification: (1) transference of genetic material and (2) expression of the target gene. The regulatory guidelines recommend that these methods should be quantitative, dose responsive, stability indicating, and well controlled.11–13,18
The main challenge for developing a potency assay for gene therapy products is their complex MoA. Gene therapy products encompass a broad range of components and strategies. They include in vivo approaches where viral vectors, plasmids, and nucleic acids are administered directly to patients. Additionally, ex vivo methods involve the modification of cells outside the body (cell-based ATMPs), which are then reintroduced into the patient. In the realm of in vivo administration, adeno-associated viruses (AAVs) and oncolytic viruses, such as Adenoviruses and Herpes Simplex Virus (HSV), are widely employed. Meanwhile, gamma-retro- and lentiviruses are predominantly utilized in ex vivo gene therapy, although they also hold potential for in vivo applications. 29 For AAV, there are two key parts to potency assays: (1) transduction and (2) quantitative readout. It is important to optimize both parts to get the most robust method.30,31 Traditionally, AAV has poor in vitro infectivity so it may require an enhancer. 32 For the biological activity method, there is a need to think when to start the potency testing because it can be long, material and resource intensive, due to the need of a matrix approach to potency.10,33
For gene therapy products, the following potency assays are recommended (Fig. 2):
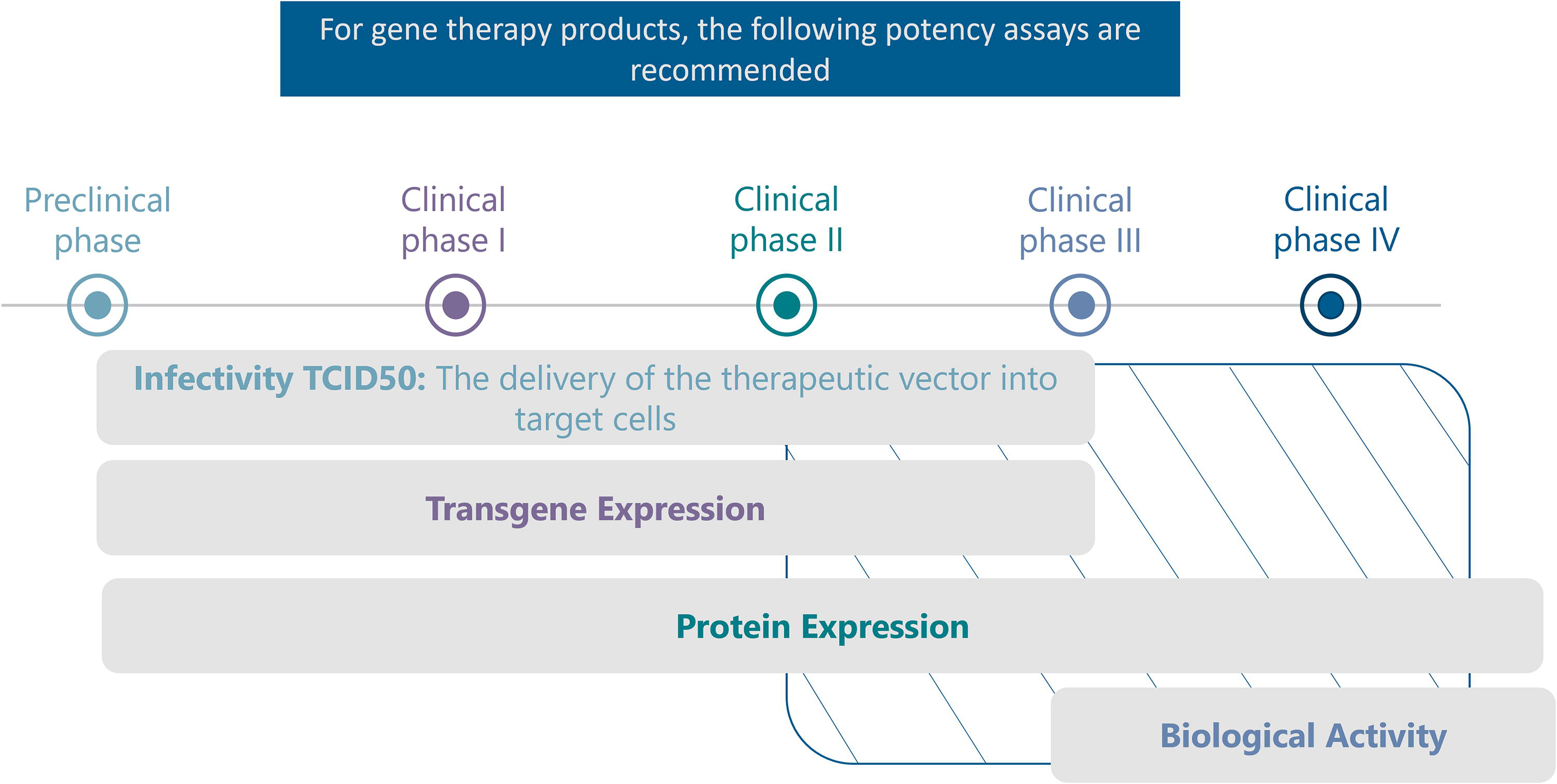
Recommendations on the type and timeline for potency assays for gene therapies. From industry feedback analysis. For some products that have been shown to be safe and effective in previous clinical trials, and that have a well-understood MoA, there is no need for phase 3 and phase 4 trials. MoA, mechanism of action.
Infectivity TCID50: 34 This assay measures the delivery of the therapeutic vector into target cells and is recommended from the preclinical phase to phase III trials.
Transgene Expression: Assessing the transcription of the Gene of Interest is essential and this assay is recommended from the preclinical phase to phase III trials.
Protein Expression: Measurement of the expressed protein helps evaluate the therapeutic potential of the gene therapy product.
Biological Activity: This assay evaluates the functional activity of the expressed Protein of Interest and is tailored to the specific product. Example: cell line screening and selection, optimize activity readout, optimize transduction, and optimize the multiplicity of infection curve, plate uniformity, and intraplate precision, prequalification. It is recommended from phase III trials to licensure application.
The ideal gene therapy potency test would simultaneously quantify the transfer of genetic material and changes in target gene expression. The one-step reverse transcription Droplet Digital PCR (RT-ddPCR) method can simultaneously measure vector integration and effect. 30 ddPCR workflow provides absolute quantification of target genes without standard curves, reducing variability. ddPCR methods are also ideal for cell therapy transgene quantification. The ddPCR assay allows for the absolute quantification of DNA copy number, but it requires purified DNA. Catalent has developed a relative potency bioassay using real-time quantitative reverse transcription (RT-qPCR) in a duplex format to assess relative transcription activity in cells treated with ligands or transgenic vectors. The assay utilizes two fluorescent dyes with minimally overlapping emission spectra that allow real-time monitoring of the gene expression of both target and normalizer genes. 30 In contrast, mRNA transcriptional assay assesses the functionality of inserted gene constructs, but it is a multistep assay, and it requires a standard curve. Finally, the traditional flow cytometry assay evaluates translational activity and determines the population of cells expressing a particular protein but can be difficult to standardize. However, all these assays are limited in their ability to indicate if a gene construct is functionally inserted.
Moreover, the presence of partial and empty AAV impurities can significantly impact product potency. Partial impurities refer to AAV particles that contain incomplete viral genomes, while empty impurities are AAV particles devoid of any genome. The manufacturing process inherently produces empty capsids, which lack the therapeutic transgene. While the impact of empty capsids on clinical outcomes remains unclear, there are concerns regarding immunogenicity and efficacy. As these capsids are considered a product-related impurity, it is crucial to control their presence during manufacturing and continuously monitor them through analytical testing.35–37 Potency assays help assessing the impact of these impurities on product potency by measuring the biological activity of the therapeutic vector. By quantifying the functional potency of the AAV product, researchers can understand how the presence of partial and empty impurities influences its efficacy. This information is critical for optimizing manufacturing processes, ensuring the production of AAV vectors with high potency and therapeutic value. In addition to impurities, the post-translational modifications (PTMs) of AAV capsids can also affect product potency.38,39 PTMs are chemical modifications that occur on proteins after translation, altering their structure and function. 40 Capsid PTMs, such as phosphorylation or glycosylation, can impact the binding, transduction efficiency, and immune response of AAV vectors.39,41–43 Understanding the influence of capsid PTMs on product potency requires comprehensive characterization and analysis. 44 Potency assays can provide insights into how these modifications affect the functional properties of AAV vectors. By evaluating the potency of modified AAV variants, researchers can determine the optimal PTM patterns for achieving desired therapeutic outcomes while ensuring stability and efficacy.
In the field of gene therapy, AAVs are widely used as vectors for delivering therapeutic genes. While traditional production methods such as triple transfection in HEK293 cells are effective, they present challenges related to high production costs, complexity in operations, and scalability. To address these limitations, alternative production approaches, including the use of insect cell lines such as Spodoptera frugiperda (Sf9), are being explored. The baculovirus expression vector system offers advantages like cost efficiency and higher AAV yields compared with traditional systems. Liu et al 45 provide a comparative analysis of recombinant AAV (rAAV) vectors produced using HEK293 mammalian cells and Sf9 insect cells across various production scales. The findings indicate that Sf9-based production yielded higher total viral genome numbers compared with HEK293, along with a superior full-to-empty capsid ratio (93.2% vs. 70.8%). Additionally, Sf9 vectors exhibited fewer DNA impurities and better aggregation profiles. Notably, the study identified more diverse PTMs, such as phosphorylation and glycosylation, in Sf9-rAAV, which may impact vector stability and biological interactions. Infectivity and transgene expression were also more favorable for Sf9 vectors in vitro, supported by tissue culture infectious dose assays and ELISA results. In vivo testing revealed comparable efficacy for both systems in a mouse model for ocular neovascularization. These results suggest that while both production systems are effective, Sf9-based approaches may offer advantages in yield, purity, and vector quality for certain gene therapy applications. However, the insect cell production platform introduces distinct analytical and potency-related challenges. The paper “Methods Matter: Standard Production Platforms for Recombinant AAV Produce Chemically and Functionally Distinct Vectors” 43 highlights how AAV production methods significantly influence vector potency, a critical factor for gene therapy. Vectors produced in HEK293 cells demonstrated superior transduction efficiency and gene expression compared with those from the baculovirus-Sf9 system, both in vitro and in vivo. This potency difference is attributed to fewer immunogenic PTMs in HEK-derived vectors and reduced host cell protein impurities. In contrast, Sf9-derived vectors showed non-native capsid modifications and higher HCP contamination, potentially impairing receptor binding and intracellular processing.
Ensuring adequate quality control is crucial to maintain the functional integrity of AAV vectors. Analytical testing for residual DNA from host cells and baculoviruses is required to limit impurities, with regulatory guidelines setting thresholds below 10 ng/dose. Additionally, detecting and preventing the presence of adventitious agents, such as the Sf-rhabdovirus, is a critical aspect of maintaining product safety. Despite the potential advantages, limitations concerning the potency of AAV vectors produced in insect cells persist. Differences in PTMs and packaging efficiency may impact the biological activity of AAV products. Further studies are necessary to optimize production parameters and validate the equivalence of insect-cell-derived vectors to those produced using mammalian cell systems, ensuring their suitability for clinical applications.
CELL THERAPY: POTENCY ASSAY CHALLENGES AND RECOMMENDATIONS
For cell therapy products, we provide these recommendations on the type and timeline for potency assays (Fig. 3):

Recommendations on the type and timeline for potency assays for cell therapies. From industry feedback analysis. For some products that have been shown to be safe and effective in previous clinical trials, and that have a well-understood MoA, there is no need for phase 3 and phase 4 trials.
Cell Viability and Proliferation Assays: These assays assess the viability and proliferation of administered cells; they are recommended to begin at the preclinical phase onwards.
Differentiation Assays: Evaluating the ability of administered cells to differentiate into the desired cell types is crucial, and these assays are recommended from the preclinical phase.
Various techniques can be employed to evaluate the extent of cell differentiation. This can include immunocytochemistry or immunofluorescence staining to detect specific markers associated with the desired cell type. Flow cytometry can also be used to analyze the expression of cell surface markers. Additionally, gene expression analysis through techniques like quantitative PCR or RNA sequencing can provide molecular evidence of differentiation.
Functional Assays: These assays evaluate the specific functions of administered cells in relation to the desired therapeutic effect. They are recommended from the preclinical phase. Immunophenotyping: Immunophenotyping assays, utilizing techniques such as flow cytometry, characterize cell surface markers or intracellular markers. Their implementation is recommended from phase III clinical trials. Cytokine or Growth Factor Secretion Assays: These assays measure the production and secretion of specific cytokines or growth factors by the administered cells. Their recommended use begins from phase III clinical trials.
Potency assays for cell therapy encounter difficulties in measuring biological activity accurately while ensuring product quality for patient safety and efficacy during manufacturing. Among the most direct measures of a T cell-based therapy’s biological activity are cell-killing assays, which quantify the molecule’s ability to induce cell death. However, these assays have limitations in serving as quality-control (QC) tools due to high assay variability and labor- and time-intensive procedures, making them impractical for sustained use from development to commercialization.46,47 To address these challenges, reporter-gene assays have emerged as promising alternatives for QC purposes. In reporter-gene assays, engineered cell lines express luciferase under the control of biologically relevant response elements for T-cell activation, such as nuclear factor of activated T cells or nuclear factor kappa B. These assays allow measurement of events upstream of cell killing, providing faster, easier, and more reproducible results compared with cell-killing assays. Consequently, they are considered more suitable for QC purposes. However, it is essential to validate that these assays accurately reflect the MoA of the cell therapy product to ensure their appropriateness for QC usage. By leveraging reporter-gene assays effectively, researchers can enhance the efficiency and reliability of potency assessment throughout the development and manufacturing of cell therapies. 47
COMBINED ATMP: POTENCY ASSAY CHALLENGES AND RECOMMENDATIONS
Combined ATMP can be gene, cell, or tissue based, but they also contain one or more medical devices as an integral part of the medicine. Regulatory agencies require robust potency assay data for the approval of combined ATMPs. However, the dynamic nature of combined ATMPs and the lack of established guidelines specific to these therapies poses specific challenges. First, they involve the integration of different components, such as cells, gene vectors, and biomaterials and devices. The potency assays need to account for the interactions between these components to accurately assess the overall therapeutic activity. Then, combined ATMPs often exhibit heterogeneity in terms of cellular composition, gene expression levels, and biomaterial properties. This heterogeneity can impact the potency assay results, making it challenging to establish consistent and reproducible measurements. In addition, potency assays should measure the relevant functional attributes of the combined ATMPs. However, identifying the most appropriate functional parameters and developing assays that capture these attributes can be challenging. The assays should reflect the desired therapeutic effect, such as cell viability, differentiation potential, gene expression, tissue integration, or target-specific activity.
An example of a combined ATMP is the technique known as “autologous chondrocyte implantation (ACI),” 48 which is employed to repair damaged joint cartilage. In this procedure, the patient’s own cells, specifically autologous chondrocytes (cartilage cells), are cultivated and multiplied within a laboratory setting until a sufficient quantity is achieved. These expanded cells are then carefully placed onto a collagen membrane, forming a construct. Subsequently, the construct is surgically implanted into the area of the damaged cartilage. The transplanted chondrocytes function to repair the impaired tissue, with the collagen membrane serving as a medical device that securely holds the cells in place, facilitating their restorative activity. MACI (matrix-induced ACI) 49 stands out from previous generations of ACI due to its streamlined and advanced approach to implanting cartilage cells. Unlike its predecessors, MACI simplifies the process by directly and uniformly applying a patient’s newly cultivated cartilage cells onto a porcine collagen membrane during manufacturing. This robust membrane can be customized to precisely match the size of the patient’s cartilage defect and is securely affixed in place during surgery using fibrin glue, eliminating the need for sutures. This technique results in a simpler and less time-consuming surgical procedure, minimizing trauma to the surrounding tissue. 50
Potency testing for MACI is based on the measurement of aggrecan (the major proteoglycan in the articular cartilage) mRNA expression by real-time PCR. The applicant is recommended to collect identity and potency data for final cell suspension for loading on the membrane and provide a report with proposed IPC (in-process controls) acceptance criteria (Table 1).
Overall, Figure 4 proposes the following recommendations for measuring the potency of combined ATMPs, where it is important to include the device in the validation process.
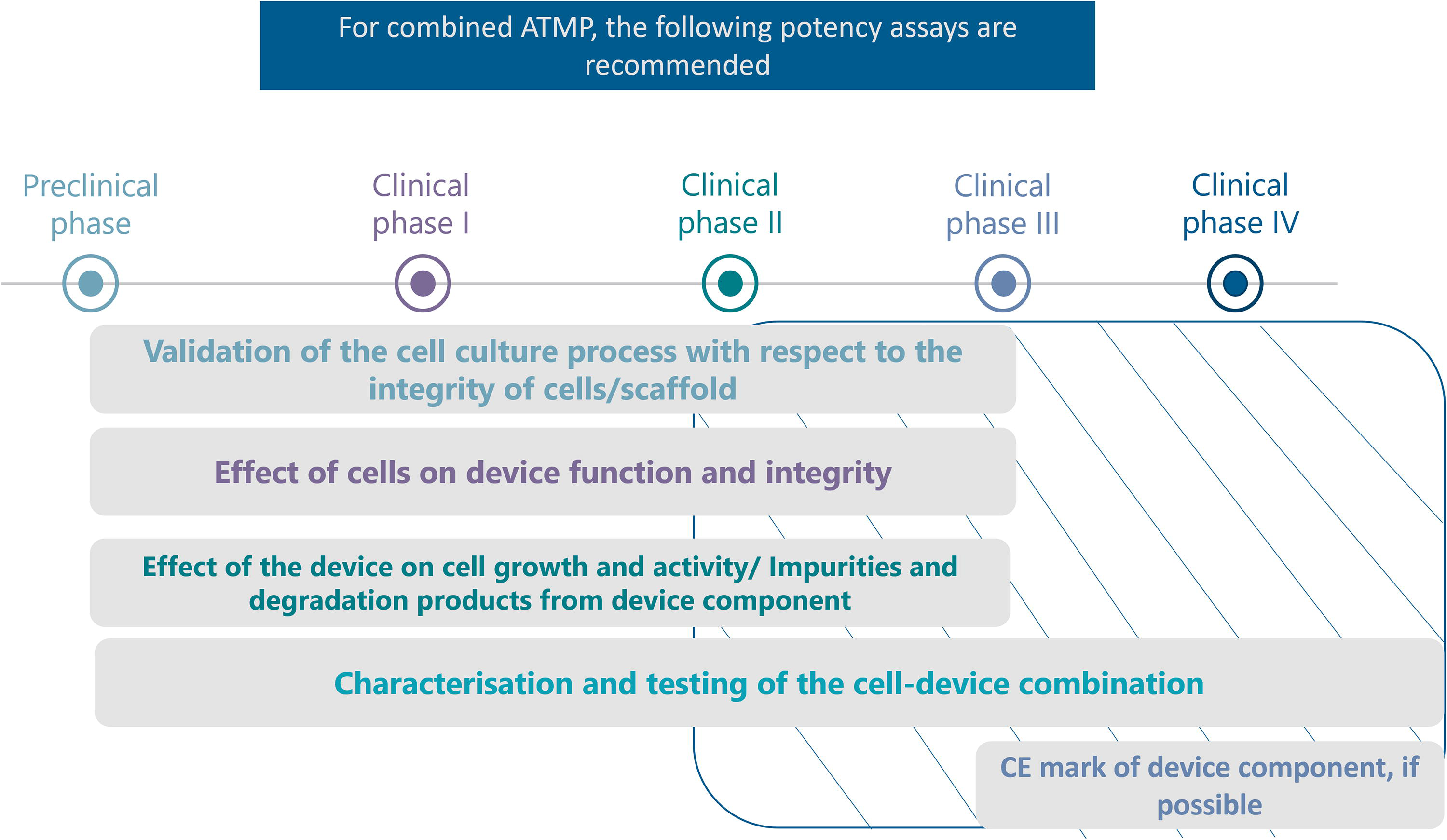
Recommendations on the type and timeline for potency assays for combined ATMP. From industry feedback analysis. For some products that have been shown to be safe and effective in previous clinical trials, and that have a well-understood MoA, there is no need for phase 3 and phase 4 trials.
RNA-BASED ATMP: POTENCY ASSAY CHALLENGES AND RECOMMENDATIONS
In the evolving landscape of RNA-based therapies, the classification of these products as gene therapies remains a complex and unsettled issue. Regulatory frameworks, particularly in the United States and European Union, offer differing perspectives. 6 The FDA’s definition of gene therapy focuses on the modification of genetic material within cells, a process that RNA-based therapies, such as mRNA drugs, do not typically involve. As a result, these therapies are unlikely to be classified as gene therapy in the United States. However, in the European Union, mRNA-based therapies may fall under the broader category of gene therapy medicinal products as defined by the EMA, especially when the mRNA acts as a recombinant nucleic acid with therapeutic effects. 51 Notably, mRNA vaccines against infectious diseases are excluded from this classification. This divergence has implications for the requirements for the Marketing Authorization to ensure patient safety and security (i.e., germline transmission, insertional mutagenesis, tumorigenicity, and embryo/fetal and perinatal toxicity). The absence of integration into the genome and the transient nature of mRNA expression further complicates the classification, indicating a need for tailored regulatory guidance that considers these unique attributes. As the regulatory landscape continues to develop, the current guidelines for gene therapies may serve as a reference point, but the distinct features of mRNA necessitate specific considerations to ensure appropriate oversight and safety evaluation. The risk of germline transmission and insertional mutagenesis is generally considered negligible for mRNA-based therapies due to the transient nature of mRNA. These risks are more associated with DNA-based gene therapies. 6
mRNA-based vaccines have advantages over other platforms, such as adaptability, manufacturing speed, and inherent adjuvant qualities that boost the immunological response. A breakthrough therapy designation by the FDA (in February 2023) 52 and a Priority Medicines scheme designation by the EMA (in April 2023) were given to the experimental personalized mRNA cancer vaccine (Moderna/Merck & Co’s mRNA-4157/V940) 53 when used in combination with the immune checkpoint inhibitor pembrolizumab (Keytruda, anti-PD1) as adjuvant therapy for patients with high-risk stage III or IV melanoma after complete resection. Recent three-year data 52 further demonstrated sustained improvement in recurrence-free survival and distant metastasis-free survival compared with Keytruda alone in these patients (NCT05933577).
Similarly, Roche/BioNTech’s RO7198457 (autogene cevumeran) is being evaluated in a phase 2 IMCODE101 trial (NCT03815058) in combination with pembrolizumab in patients with previously untreated advanced melanoma. Additionally, BioNTech announced positive topline results from a phase 2 trial of BNT111, another mRNA immunotherapy candidate, in patients with advanced melanoma, showing promising efficacy. Other very encouraging results have also emerged from a phase 1 trial (NCT04161755), wherein RO7198457 efficacy was assessed in combination with anti-PD-L1 immune checkpoint inhibitor atezolizumab and chemotherapy in patients with resected pancreatic ductal adenocarcinoma, 54 one of the deadliest cancer types. Three-year follow-up data from this trial indicated persistent immune response and delayed tumor recurrence in some patients. 55
Potency testing of mRNA products presents several challenges, as discussed during the EMA Conference on RNA-based medicines in 2023. 56 Key issues include: 56 (1) The need for highly sensitive techniques for more potent vaccines with potentially lower doses, especially with developments like self-amplifying mRNA (sa-mRNA). Ongoing research and development are expected to improve these techniques; (2) The potential lack of specific detection antibodies against each of the antigens in a multivalent vaccine product. This challenge could be addressed by combining a number of biophysical approaches, including Surface Plasmon Resonance, to enhance detection capabilities; and (3) The potential risk of cross-reactivity of antibodies used to detect multivalent products. This challenge could be managed by using antibodies labeled with different fluorophores, as long as labeling does not affect their binding affinity for the expressed antigen.
While drawing on knowledge from traditional gene therapy research, mRNA therapeutics present unique challenges and require tailored strategies for optimization. Evaluating mRNA-based therapies involves careful consideration of several aspects. These include assessing the efficiency of mRNA uptake, intracellular processing, delivery mechanisms, and stability within the cytoplasm-factors that differ from traditional gene therapy approaches. Additionally, it is crucial to evaluate the efficiency of translation, the duration, and magnitude of protein expression, and how these are influenced by mRNA degradation over time. Establishing a dose-response relationship between mRNA concentration and resulting protein expression is essential to identify the minimum effective dose without causing toxicity. Lastly, evaluating the immunogenicity of mRNA molecules, including inflammatory cytokine production and immune cell activation, is necessary for understanding potential immune responses.
DISCUSSION
Approved potency assays for ATMPs exhibit a complex relationship with scientific advancements and evolving regulatory guidance. While some consistency exists, with assays acknowledging inherent variability through multipronged approaches and demonstrating correlation with animal models, a gap remains. Many approved assays rely on established techniques like flow cytometry, viability assays, and reporter gene assays. These techniques, while valuable, may not fully capture the complexities of novel ATMPs with advanced mechanisms of action. Newer innovations, such as 3D cell cultures or microfluidic platforms that better mimic the in vivo environment, are under exploration but haven’t been widely adopted in approved assays yet. This inconsistency is further amplified by the lack of standardized reference materials and universal assays across different ATMP types. Additionally, for some therapies, the most relevant functional attributes for potency measurement are not fully understood. These inconsistencies likely stem from the rapid evolution of the ATMP field, the initial focus on safety in early development stages, and the challenge of validating new technologies. However, the new FDA guidance, with its emphasis on a risk-based approach, offers a path forward. By encouraging early adoption of innovative methods, continuous improvement, and development of product-specific solutions, this approach can bridge the gap between approved assays and scientific advancements, ultimately leading to more robust and informative assays for next-generation ATMPs.
It is important to note that while the guidance provides extensive direction on potency assurance for CGTs, certain aspects related to Advanced Therapies are not yet explicitly addressed. Various technologies and platforms utilized in ATMPs, such as gene editing techniques (e.g., CRISPR/Cas9) or distinct viral vectors, may have each unique potency considerations. For example, with CRISPR/Cas9, potency considerations may include the efficiency of gene editing, off-target effects, and the persistence of desired genetic modifications. Additionally, ATMPs often involve personalized therapies, tailored to individual patient characteristics. Today there is no in-depth guidance on addressing potency concerns within the context of personalized medicine. For example, how can variability in patient-specific factors, such as genetic background and disease state, be accounted for in potency assessments? And how can real-time monitoring or adaptive strategies be integrated into potency assessment protocols to optimize personalized treatment regimens? The intricacies of potency assessment for tissue-engineered products are not extensively covered, considering the complex interactions involved. Assessing potency for tissue-engineered products involves understanding their integration within the host environment and characterizing the dynamic interactions with surrounding tissues and incorporated molecules. Challenges include quantifying spatial organization and addressing scalability issues, particularly in personalized therapies. As the field of ATMPs evolves, emerging technologies may rise new challenges.
The integration of artificial intelligence (AI) in CGT and ATMP development offers promising avenues for data analysis, predictive modeling, and process optimization, though its role in potency testing remains largely unexplored. AI-driven applications, such as nondestructive quality assessments through cell imaging for viability analysis and robotics-assisted procedures like laser optoporation, are emerging as innovative tools in manufacturing. Additionally, machine learning can assist in predicting treatment outcomes, identifying optimal patient populations, and improving manufacturing workflows. Despite its potential, AI adoption faces regulatory hurdles, as neither the FDA nor EMA has issued specific guidelines for its use or validation in potency testing within GMP environments, signaling an area requiring future exploration and standardization.
CONCLUSION
In conclusion, the product development journey of CGT/ATMP still raises several challenges, particularly in potency assay design. Based on existing guidelines and data from both industry and literature, we propose a product-specific approach framework including the latest scientific innovation approaches. By addressing technical and regulatory challenges and advocating for tailored approaches, this framework aims to facilitate the safe and effective production of ATMPs, ultimately advancing patient care in areas with high unmet medical needs. In the future, guidance for ATMP potency assays should prioritize standardization across diverse product types, define acceptable assay variability, and establish clear links between in vitro potency data and clinical outcomes. We highlight the need for ongoing refinement and adaptation of potency assurance strategies as the field of CGTs/ATMPs evolves. Factors such as patient-specific variability, immune interactions, and long-term potency maintenance require continuous attention and refinement of potency assays and regulatory guidelines. Furthermore, addressing emerging technologies such as AI, considering pediatric-specific assessments, and adapting to real-world data will also be essential to ensure the effectiveness and safety of ATMPs.
AUTHORS’ CONTRIBUTIONS
A.A. conceived and designed the study, collected, and analyzed the data, interpreted the results, and drafted the article. M.B.J. contributed to the study design, data collection, and analysis related to RNA therapies as well as provided critical revisions for intellectual content. A.Z. provided feedback on the article and approved the final version for publication. G.B. provided expertise in specific aspects of the study design and article preparation, critically revised the article for intellectual content, and reviewed and approved the final version of the article. All authors have read and approved the final article and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no conflicts of interest relevant to this work.
FUNDING INFORMATION
This work was funded by ProductLife Group.