
Abstract
Duchenne muscular dystrophy (DMD) is a severe, progressive genetic disorder primarily affecting boys, characterized by muscle degeneration due to mutations in the DMD gene encoding dystrophin, a crucial protein for muscle fiber integrity. The disease leads to significant muscle weakness and eventually to loss of ambulation. Adeno-associated viral (AAV)–microdystrophin (MD) gene therapy shows promise in preclinical and clinical settings. However, muscle fibrosis, a consequence of chronic inflammation and extracellular matrix remodeling, exacerbates disease progression and may hinder therapeutic efficacy. Periostin, a matricellular protein involved in fibrosis, is upregulated in DMD rodent models and correlates with collagen deposition. We previously developed an antisense oligonucleotide strategy to induce exon 17 skipping and so reduce periostin expression and collagen accumulation in the fibrotic D2.mdx mouse model of DMD. Here, we investigated the combined effects of periostin modulation and AAV-MD1 treatment. We found that systemic periostin splicing modulation significantly improved muscle function, assessed by forelimb grip strength and treadmill performance. Importantly, periostin exon skipping increased the MD protein expression. These findings suggest that targeting periostin in conjunction with MD therapy could represent a valid therapeutic strategy for DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe, progressive genetic disorder characterized by muscle degeneration and weakness. It affects approximately 1:5,000 live male births worldwide 1 and is caused by mutations in the DMD gene, which encodes dystrophin, a crucial protein for muscle fiber integrity. 2 Loss of ambulation occurs by the early teenage years and, without intervention, cardiac 3 or respiratory failure 4 cause death in early adulthood.
Adeno-associated viral–microdystrophin (AAV-MD) gene therapy stands as one of the most promising avenues for treating DMD. We developed AAV8-Spc5-12-MD1, a viral vector presently undergoing clinical trials (GNT0004) 5,6 that expresses microdystrophin-1 (MD1). MD1 is delivered to the skeletal muscles by the AAV8, which have proved widespread and robust transgene expression in skeletal muscles tissues 7 and its expression is under the control of the synthetic muscle-specific promoter Spc5-12. 8 This shorter version of dystrophin protein showcases robust protein expression in murine and canine DMD models when delivered through AAV vectors, and is associated with functional recue. 9 –13 An AAV-MD clinical product has received FDA approval and is now available in the market (Elevidys®). 14 While promising, published clinical data suggest that this approach still requires optimization.
In DMD, muscle fibers degeneration triggers chronic inflammation, leading to the excessive deposition of extracellular matrix (ECM) proteins. This fibrotic tissue deposition which exacerbates muscle weakness, is a major pathological feature of DMD and significantly contributes to disease progression. 15
Periostin is a matricellular protein that plays a critical role in tissue repair, inflammation, and fibrosis. 16 It promotes collagen deposition and fibrotic tissue formation in the skeletal muscle. 17 –19 Indeed, we recently showed that periostin protein expression is positively correlated to the collagen I and III deposition in the mdx mouse diaphragm muscle. 17 Consistently, periostin expression is significantly upregulated in the mdx gastrocnemius and diaphragm muscles 20 and in the mdx-4cv diaphragm muscles, 18 two mouse models of DMD characterized by degeneration/regeneration cycles.
Alternative periostin isoforms are generated by alternative splicing of exons 16–23 region 21 with the expression of isoforms containing exon 17 being significantly increased in the diaphragm muscle of muscles of D2.mdx mouse compared to the healthy DBA/2J littermate controls. 19,22
We recently showed that an oligonucleotide antisense strategy, based on periostin exon 17 skipping ameliorated the dystrophic pathology observed in D2.mdx mice. 22
Here, we combined the periostin exon 17 skipping with intramuscular AAV-MD delivery. The periostin exon 17 skipping significantly improved muscle function, as assessed by running treadmill and forelimb grip strength while it failed to protect the tibialis anterior (TA) muscle from repeated eccentric contractions. Notably, MD protein was significantly expressed by the AAV-MD1 treatment in the TA where it protected the muscle from repeated eccentric contractions damage. These data suggest that the combination of these treatments could provide an additive protection and functionality to muscle.
METHODS
Chronic vivo-PMO-postn intraperitoneal injections
All animal procedures were performed in accordance with UK government regulations and were approved by the UK Home Office under Project License P36A9994E. Ethical and operational permission for the in vivo experiments was granted by the Animal Welfare Committee of Royal Holloway University of London. Two-week-old dilute brown non-agouti (DBA)/2J wild-type (WT) and D2.mdx mice were randomized by body weight into groups of four to six mice. Mice received weekly intraperitoneal (IP) injections of 10 mg/kg scramble vivo-phosphorodiamidate morpholino oligomer (PMO) (5′−3′: CCTCTTACCTCAGTTACAATTTATA) (vivo-PMO-scr) or periostin exon 17 skipping vivo-PMO (5′−3′: CTTCCGTTTTGATAATAGGCTGAAGACT) (vivo-PMO-postn) from 2 to 12 weeks of age. vivo-PMO-scr and postn (Genetools, USA) were dissolved in 0.9% NaCl solution. The dose was adjusted weekly based on the individual body weight.
Production of AAV-MD1
Previously, murine-specific MD1 cDNA sequences, optimized for mRNA expression, were developed. 9 The MD1 construct consists of a truncated dystrophin sequence, with deletions in the spectrin-like repeat domain (exons 4–23) and the C-terminal (CT) domain (exons 71–78). This sequence ends with the final three amino acids from exon 79, followed by three stop codons, and includes the SV40 polyadenylation signal. To enhance translation initiation, we modified the Kozak sequence from TCAAAATGC to CCACCATGC. Furthermore, the untranslated regions (UTRs) of the dystrophin gene were removed to reduce the length of the inverted terminal repeat (ITR) sequences flanking the MD cassette. The expression of the construct is controlled by the highly active muscle-specific synthetic promoter SPc5-12. 8 Optimization of the cDNA sequences for murine expression was performed by GENEART (Regensburg, Germany). The finalized construct was then excised from the pGA4 vector and inserted into a pAAVITR-based plasmid, which contained the 360-bp SPc5-12 promoter, an SV40 polyadenylation signal, and the AAV serotype 2 ITRs. The MD1 cDNAs were 3606 bp in length, and the total transgene cassette sizes (including SPc5-12, SV40 poly A, and ITRs) for AAV-MD1 were approximately 92% of the WT AAV2 genome length (4682 bp).
HEK293T cells were cultured in roller bottles in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and incubated at 37°C with 5% CO2. All cell culture reagents were obtained from Invitrogen. Pseudotyped recombinant AAV2/9 vectors were produced by calcium phosphate coprecipitation and triple transfection using the AAV2ITR-MD1 construct, along with the pAdΔF6 and pAAV5E18-VD2/9 plasmids, at a molar ratio of 1:1:1 in HEK293T cells. After 72 h, cells were lysed, and viral particles were purified through iodixanol step-gradient ultracentrifugation. The vector genomes (vg) were quantified using dot blot hybridization.
Intramuscular injection of AAV-MD1
At 6 weeks of age, mice were anesthetized with 2–4% isoflurane and injected with AAV8-Spc5-12-MD1 vector 10 (low dose: 1e + 10 vg/30 µL/muscle; high dose: 4e + 10 vg/30 µL/muscle) in both TA muscles. Control mice received NaCl (30 µL). Mice were euthanized at 13 weeks of age for tissue collection.
Forelimb grip strength
Forelimb grip strength was measured at 12 weeks using a grip strength meter (Linton Instrumentation, Palgrave Diss, Norfolk, UK). Mice were gently placed on a grid and their tail was pulled until they released their grip. This procedure was repeated five times, with the highest force recorded, normalized to body weight, and averaged. Measurements were performed every two days for consistency.
Running treadmill
At 12 weeks, mice were tested for fatigue resistance using a Treadmill Simplex II (Columbus Instrumentation, Columbus, Ohio, USA) set at a 15% incline. After a 5-min acclimatization, the treadmill speed started at 5 m/min and increased by 0.5 m/min every minute until exhaustion. The distance run before failure was calculated based on the recorded time to fatigue.
In situ tibialis anterior muscle electrophysiology
At 13 weeks, under deep anesthesia (Dolethal 50 mg/kg, Buprenodale 75 µg/kg), TA muscle functionality 10,23 (TREAT-NMD SOP DMD M.2.2.005) was assessed by stimulating the common peroneal nerve. Maximum isometric tetanic force (Po) was recorded at varying stimulation frequencies (10–180 Hz). Specific force (N/cm2) was calculated by dividing Po by muscle cross-sectional area. Muscle resistance to eccentric contractions was evaluated by lengthening the muscle after 150 Hz stimulation for 700 ms, followed by a 15% lengthening at 0.75 Lo/s.
Tissue collection and storage
Post-electrophysiology, mice were euthanized by cervical dislocation. TA muscles, diaphragm, and heart were harvested, weighed, and either embedded in OCT medium (VWR, Lutterworth, UK) for histology or snap-frozen for RNA/protein analysis. All samples were stored at −80°C.
RNA extraction, cDNA synthesis, and qPCR
Muscle tissue (30 mg) was homogenized in RLT buffer (QIAGEN, Hilden, Germany) and RNA was extracted using the RNeasy Fibrous Tissue Mini Kit (QIAGEN, Hilden, Germany). RNA concentration and purity were assessed via Nanodrop spectrophotometry. cDNA was synthesized using the QuantiTect Reverse Transcription Kit (QIAGEN, Hilden, Germany). qPCR was performed on the LightCycler480 system (Roche, Mannheim, Germany) with optimized primers (Supplementary Table S1). Gene expression was normalized to Ribosomal Protein Lateral Stalk Subunit P0 (Rplp0). 24
Protein extraction and Western blot
Proteins were extracted from frozen muscle tissues using RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was determined using the BCA protein assay. Equal amounts of protein (50 µg) were separated on 12 well 3–8% Tris Acetate (dystrophin) or 4–12% Tris Bis (periostin) gels with their respective running buffers for 1.5 h at 150 V. Each gel contained the protein ladder (Hi Mark for dystrophin, Chameleon for periostin) and appropriate positive and negative controls. Post electrophoresis, electro transfer onto nitrocellulose membranes was prepared, each cassette containing two gels for transfer. Electro transfer was run at 30 V for 2 h for both proteins. After transfer, ponceau staining was used to establish successful transfer before removal and blocking for 1 h. Primary antibodies (dystrophin, #mannex1011c, DSHB, Iowa City, Iowa, US, 1:100; periostin, #AF2955; R&D Biosystems, Minneapolis, Minnesota, USA, 1:2000) were then added for overnight incubation at 4°C. α-tubulin (#ab4074, Abcam, Cambridge, UK, 1:2500) was used as housekeeping for both proteins.
The following day, the membranes were washed and incubated with secondary antibodies conjugated to 680 or 800 nm fluorophores (Licor, USA) and scanned using the Odyssey CLx system (Licor, Lincoln, Nebraska, USA) and analyzed on Image Studio lite 5.2. Relative protein concentration was determined by normalization of the protein of interest to the housekeeping α-tubulin comparatively across samples.
Cryosectioning and immunofluorescence staining
Muscle tissues embedded in OCT were cryosectioned (10 µm) at −20°C using a cryostat. For dystrophin immunofluorescence, muscle sections were fixed, incubated with primary antibodies (dystrophin, #mannex1011c, DSHB, Iowa City, Iowa, USA; laminin, 1:300, #ab11575, Abcam, Cambridge, UK), followed by secondary antibodies (Alexa 488 and streptavidin-568), thanks to the Mouse on Mouse immunodetection kit (#BMK-2202, Vector Laboratories, Newark, California, USA). Nuclei were stained with DAPI. For Sirius Red staining, muscle sections were incubated in 0.3% Sirius Red solution, dehydrated, and mounted with DPX mounting solution.
Image acquisition and analysis
Immunofluorescence and histological images were captured at 20× magnification using a Nikon NiE Upright microscope (Nikon Corporation, Tokyo, Japan). Dystrophin-positive fibers and Sirius Red-stained areas were quantified using Fiji software. The centro-nucleated fibers percentage and the whole muscle section area were analyzed using the MuscleJ plugin on Fiji. 25
Hydroxyproline quantification
Hydroxyproline levels in DIA muscle samples were determined using the Sigma–Aldrich Hydroxyproline Assay Kit (MAK357). Tissue samples were initially homogenized in ultrapure water, then mixed in equal volumes with 10 M NaOH and heated at 100°C for 2 h. After cooling on ice, the samples were neutralized with 10 M HCl, vortexed, and centrifuged. A 10 µL aliquot of each neutralized hydrolysate was placed in duplicate in a clear, flat-bottomed 96-well plate along with hydroxyproline standards and evaporated at 65°C. Following evaporation, 100 µL of oxidation reagent was added to each well and incubated at room temperature for 20 min. Then, 50 µL of developer solution was added and incubated at 37°C for 5 min, followed by the addition of 50 µL of DMAB concentrate and a final incubation at 65°C for 45 min. Absorbance was measured at A560 nm, and hydroxyproline concentrations were calculated by comparison to the standard curve.
Statistical analysis
Data were expressed as mean ± SEM. Outliers were identified using ROUT analysis (Q = 1%) and excluded. Statistical comparisons were performed using one-way or two-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. A p value <0.05 was considered statistically significant. We only presented the comparisons between vivo-PMO-postn and relatives vivo-PMO-scr groups on the graphics. The overall relevant statistics can be found in Supplementary Table S2.
RESULTS
vivo-PMO-postn improves muscle function of the D2-mdx mouse treated with intramuscular AAV-MD1
10 weeks of IP weekly injections of 10 mg/kg of vivo-phosphorodiamidate morpholino oligomer (PMO)-periostin (vivo-PMO-postn) or vivo-PMO-scramble (vivo-PMO-scr) were performed in dystrophic D2.mdx and littermate control dilute brown non-agouti (DBA)/2J (WT) mice from 2 weeks of age. Four weeks after the first injection of vivo-PMOs, 1E + 10vg (Low dose) or 4E + 10vg (High dose) of AAV-Spc5-12-MD1 were injected in TA muscles. Samples were harvested at week 13 of age. Vivo-PMO treatments were well tolerated by the mice as the weekly body mass measurement showed similar weight gain compared to WT mice (Fig. 1A).

vivo-PMO-postn improves muscle function of the D2-mdx mouse treated with intramuscular AAV-MD1.
At week 12 of age, before harvesting samples, treadmill running and forelimb grip strength tests were performed. D2.mdx treated with vivo-PMO-scr and IM-NaCl ran less distance (−38%, **p = 0.0072) (Fig. 1B) and provided less forelimb grip strength (−46%, ****p < 0.0001) (Fig. 1C) than the healthy mice. Treatment with vivo-PMO-scr alone restored the running distance (Fig. 1B) and the forelimb grip strength (Fig. 1C) to the level of the WT mice values. On these functional parameters, the low dose of IM-AAV-MD1 alone did not produced any improvement compared to the untreated D2.mdx mice (Fig. 1B–C). The high dose of IM-AAV-MD1, by itself, did not modify the running distance compared to the untreated D2.mdx mice (Fig. 1B) but significantly increased the forelimb grip strength (+53%, **p = 0.004) (Fig. 1C). The combination of vivo-PMO-postn and low dose of IM-AAV-MD1 has no effect on the running capacity (Fig. 1B) but significantly increased the forelimb grip strength only when comparing with the untreated D2.mdx (+115%, ****p < 0.0001) (Fig. 1C). The mice treated with the high dose of IM-AAV-MD1 combined with vivo-PMO-postn ran longer (+55%, *p = 0.0161) (Fig. 1B) and developed more forelimb force (+41%, **p = 0.0018) (Fig. 1C) compared to the mice treated with the high dose of IM-AAV-MD1 alone.
Then, we focused on the TA function using in situ muscle electrophysiology. TA maximal specific force (at 180 Hz) was not modified by genotype or treatments (Fig. 1D). Untreated D2.mdx mice were subjected to progressive loss of TA muscle force after eccentric contractions, indicating the fragility of the sarcolemma (Fig. 1E). While vivo-PMO-postn on its own did not procure any protection against eccentric contraction induced damage, the four groups of mice treated with AAV-MD1, regardless the dose or the combination with vivo-PMO-postn or without, showed the same improvement in membrane resistance, compared to the untreated D2.mdx mice (Fig. 1E).
vivo-PMO-postn increases MD1 protein expression but does not enhance tibialis anterior muscle histology of the D2-mdx mouse treated with intramuscular AAV-MD1
By western-blot, we observed that MD protein was brought by both dose of AAV-MD1 injected intramuscularly (Fig. 2A). Interestingly, the amount of MD protein generated by the high dose of AAV-MD1 was further increased by the combination with the vivo-PMO-postn compared to the high-dose-AAV-MD1 alone (fold change x6.78, ****p < 0.0001) (Fig. 2C). This observation was not made with the low dose of AAV-MD1 (Fig. 2C). Dystrophin immunostaining (Fig. 2B) unraveled a higher percentage of MD positive fibers with the high dose AAV-MD1 compared to the low dose of AAV-MD1 (+69%, ***p = 0.0003), demonstrating a dose effect (Fig. 2D). Nevertheless, no improvements were obtained by the combination with the vivo-PMO-postn treatment (Fig. 2D).

vivo-PMO-postn increases MD1 protein expression but does not enhance tibialis anterior muscle histology of the D2-mdx mouse treated with intramuscular AAV-MD1.
To assess the histopathological improvement, we performed laminin and nucleus (DAPI) immunofluorescence staining on TA muscle sections (Fig. 2B). Both the whole muscle cross-sectional area and normalized mass (to body weight) of the TA treated with the combination of vivo-PMO-postn and low-dose-AAV-MD1 were reduced compared to muscles treated with low-dose-AAV-MD1 alone (respectively −29%, ***p = 0.0002 and −22%, **p = 0.0015) (Fig. 2E–F). On the contrary the percentage of centrally nucleated fibers did not change after treatments and stayed higher in all the D2.mdx mice when compared to the WT—NaCl mice (Fig. 2G).
As periostin is a key pro-fibrotic actor in skeletal muscle, we next explored whether the vivo-PMO-postn treatment affected fibrosis in TA. The Sirius red staining highlights the collagen I and III in red (Fig. 2B). D2.mdx mice presented a higher collagen I and III content compared to the WT—NaCl mice (Fig. 2H). None of the treatments were able to significantly reduced the TA fibrosis (Fig. 2H). Similar observations were made when looking at the total collagen content of the TA muscle (Fig. 2I).
vivo-PMO-postn is efficient in diaphragm of the D2-mdx mouse treated with intramuscular AAV-MD1 but not in TA or heart
We performed immunoblot and qRT-PCR to study the periostin changes after the PMO. Interestingly, we observed a significant decrease in the full-length periostin isoform protein expression, which contained the exon 17, by vivo-PMO-periostin when combined with the low dose of AAV-MD1 (Fig. 3A and B). Besides, no difference in periostin mRNA was observed for both the full-length isoforms or the isoform excluding exon 17 (Fig. 3C and D). These results suggest that the treatment has only limited effect in TA muscles.
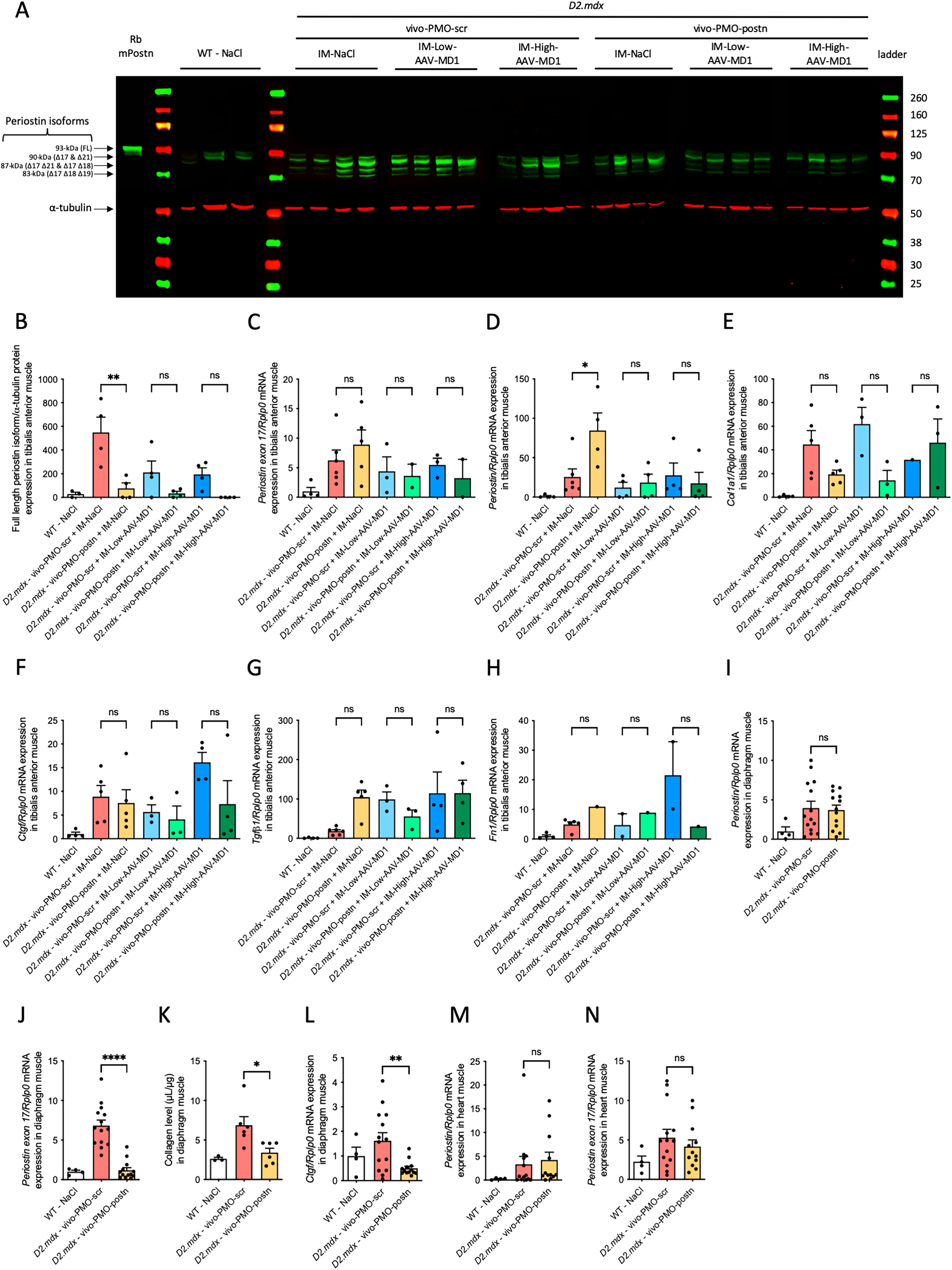
vivo-PMO-postn is efficient in diaphragm muscle of the D2-mdx mouse treated with intramuscular AAV-MD.
Next, we analyzed the mRNA of key fibrotic actors like Col1a, Ctgf, Tgf-b1, Fn1 (Fig. 3E–H). Overall, vivo-PMO-postn and AAV-MD1, by their self or in combination, were not able to reduce mRNA expression of any of these fibrotic markers (Fig. 3E–H).
Afterwards we analyzed the effect of vivo-PMO-postn treatment in other muscles. In agreement with our recent published findings 22 in diaphragm muscle, vivo-PMO-postn treatment did not change the total periostin mRNA expression (Fig. 3I) but reduced the periostin exon 17 mRNA expression specifically (fold change x-5.63, ****p < 0.0001) (Fig. 3J). Interestingly, collagen level, assessed by hydroxyproline assay, was reduced by vivo-PMO-postn treatment (−50%, *p = 0.024) (Fig. 3K), consistently with qRT-PCR quantification of the fibrotic marker, Ctgf (−69%, **p = 0.008) (Fig. 3L). Total periostin and exon17 containing mRNA expression were not modified in heart muscle (Fig. 3M and N).
DISCUSSION
In this study, we developed a protocol involving 10 weeks of vivo-PMO-postn systemic injections combined with AAV-MD1 administered intramuscularly in the TA muscle.
Intramuscular injection of AAV-MD was previously shown to be beneficial in the mdx mouse model of DMD generated under the BL10 background. 26 –28 Compared to the mdx mouse, the D2.mdx is characterized by an important muscle fibrosis that reflects more the pathophysiology of human DMD. 29,30 Therefore, it can be considered as a better mouse model to test the combination of MD restoration and anti-fibrotic periostin exon skipping strategy. We recently showed that the beneficial effects of AAV-MD1 were also observed in D2.mdx mice, with a single injection of 4e + 12 vg AAV-MD1 at 6 weeks of age in the tail vein. 19 Here, we performed an intramuscular injection of either a low dose (1e + 10 vg) or high dose (4e + 10 vg) of AAV-MD1. Both doses were able to restore a significant amount of MD as detected by western-blot and immunofluorescence. Importantly, if no difference were observed at the total MD protein expression, we noticed more dystrophin positive fibers with the high dose of AAV-MD1 compared to the low dose, indicating a dose effect. The rationale of using a lower dose of AAV was to avoid saturation of the muscle with MD1, hence the possibility for the vivo-PMO-postn to play an additional role. Consequently, the IM-Low-Dose-AAV-MD1, by itself, and among all the parameters that we analyzed, only offered a significant protection against eccentric contractions damages, compared to the untreated D2.mdx mice. Compared with the IM-Low-Dose-AAV-DM1, the IM-High-Dose-AAV-MD1 treatment, on its own, provided some additional benefit as forelimb grip strength was restored to the WT mice level. Overall, this highlights that the AAV8-spc5-12-MD1, with these doses and administration route moderately improves the functionality of D2.mdx TA muscle, fulfilling the objective of moderate improvement that can then be further enhanced by vivo-PMO-postn treatment.
The POSTN gene undergoes alternative splicing, producing multiple isoforms with distinct functional properties (Supplementary Fig. S1). Among these, isoforms containing exon 17 (e17+) are strongly implicated in promoting fibrosis, enhancing collagen deposition and contribute to pathological ECM remodeling, as we have observed in DMD previously. 19,22 Since the D2.mdx mouse displays accelerated fibrosis, the exon 17 containing isoform is highly expressed. To investigate this isoform hypothesis further in the current study, vivo-PMO-postn IP injections were applied following the same protocol of our previous study 22 with 10 mg/kg weekly IP injections from week 2 to week 12 of age. Periostin exon 17 skipping was highly efficient in diaphragm muscle, in line with what we observed previously. 22 Based on the known functionalities of the different periostin isoforms, 19,22 this represents a shift from the fibrotic isoform to non-fibrotic isoform with vivo-PMO-postn in the diaphragm muscle. Consistently, the fibrotic marker Ctgf mRNA expression and the collagen content were reduced by the vivo-PMO-postn treatment in the diaphragm muscle. On the other hand, vivo-PMO-postn showed only a limited effect in TA. Indeed, while the periostin mRNA was not modified by the exon-skipping strategy in the TA, the full-length periostin protein was less expressed after the vivo-PMO-periostin. Consistently, the TA collagens I and III content were not reduced by the vivo-PMO-postn. This difference in efficacy for vivo-PMO-postn treatment could be explained by the different levels of fibrosis which impact TA and heart (low fibrosis), and diaphragm (high fibrosis) muscles of this model 29,30 which displays a different periostin isoform expression pattern. 19,22 Despite this restricted efficacy to the diaphragm muscle, vivo-PMO-postn treatment rescued the impairment of running capacity and the forelimb grip strength in the D2.mdx mice. Further studies would be required to identify other potential muscles and organs that could contribute to the overall muscle function benefits driven by the vivo-PMO-postn, in addition to the improvement in diaphragm function. Other anti-fibrotic treatments like Givinostat, showed the ability to reduce muscle fibrosis 31 in line with improved muscle function in DMD boys, 32 underlying the clinical validity of such strategies.
In our study we combined for the first time two genetic strategies that have previously used independently to improve muscle state and function in the D2.mdx mouse model. 19,22 The combination of vivo-PMO-postn with the low dose of AAV-MD1 did not provide any benefits in the TA muscle compared to the low dose of AAV-MD1 used alone. Even worse, this combination significantly led to TA muscle cross-sectional area reduction in line with TA atrophy. On the other hand, the combination of vivo-PMO-postn with the high dose of AAV-MD1 significantly increased the AAV-mediated MD protein expression in the TA. As this could not be explained by any fibrosis reduction, further studies would be required to unravel the non-canonical mechanisms of action vivo-PMO-periostin in skeletal muscle pathophysiology. The AAV-MD1 increase was not associated with an increase of the proportion of dystrophin positive fibers. Furthermore, we observed that the combination of high dose AAV-MD1 and vivo-PMO-postn, normalized the running capacity as well as the forelimb grip strength, compared to the high dose AAV-MD1 treatment alone. However, the treadmill and forelimb grip strength improvements obtained by the vivo-PMO-postn + AAV-MD1 were not significantly different to the ones driven by the vivo-PMO-postn alone, suggesting that these positive effects were mainly driven by the vivo-PMO-postn.
Overall, our results support the rational of combining antifibrotic treatments with strategies restoring dystrophin expression.
Footnotes
ACKNOWLEDGMENT
The authors thank Emma Popescu and Claire Gregory for technical help as well as Dr. Penelope Smith and Rob Prouse for logistic support.
AUTHORS’ CONTRIBUTIONS
L.P. and A.M. designed the study. J.T., A.B., N.L., J.M., and A.M. generated and analyzed the data. A.B. and A.M. wrote the first draft of the article. All the authors participated to the writing and validated the submitted version.
AUTHOR DISCLOSURE
The authors declare no competing interests.
FUNDING INFORMATION
This work was funded by a MDUK grant # 22GRO-PG12-0588 awarded to Linda Popplewell. Dr Alexis Boulinguiez was supported by an AFM-Telethon postdoctoral fellowship and internal pump-prime funding from Royal Holloway, University of London.
SUPPLEMENTARY MATERIAL
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.