
Abstract
Multiple myeloma (MM) is an incurable hematological malignancy of plasma cells. Myeloma cells interfere with hematopoietic activities of the bone marrow, often leading to anemia, and can cause the bones to develop osteoporotic and lytic lesions. Clinical experience with chimeric antigen receptor T-cell (CAR-T) therapy targeting B-cell maturation antigen (BCMA) has been promising, with good response rates, favorable safety profiles, and low incidences of severe cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome. However, CAR-T therapy in MM is accompanied by several new challenges, including therapeutic failure and relapse, and much attention has been paid to the further development of B-cell maturation antigen-chimeric antigen receptor (BCMA-CAR). Although most of the reported benefits of BCMA-CAR have been discussed, whether cancer can be eliminated, as well as the efficacy of CAR-T therapy for anemia and bone lesions, both myeloma-defining events, have not yet been reported in any animal model. In this study, we designed and verified a novel BCMA-specific chimeric antigen receptor (CAR). Our BCMA-CAR demonstrated the fundamental properties of CAR-T cells, including target-specific cytotoxic activity, cytokine production, and in vivo antitumor effects. In addition, we evaluated the therapeutic effect of BCMA-CAR in mice by imaging bone lesions and conducting blood examinations. Tumor mouse models showed systemic progression of MM in the bone marrow, and mice treated with saline or nongene modified T cells showed continued tumor progression, progressive bone lesions, and prolonged anemia. In contrast, all mice treated with gene modified T cells achieved a complete response, improved anemia to the level observed in normal mice, and suppressed progression of bone lesions. We concluded that anemia was improved with BCMA-CAR-T cell therapy. However, novel strategies to support the recovery of bone lesions by enhancing CAR-T cell function must be developed.
INTRODUCTION
The development of novel therapies for multiple myeloma (MM) has progressed substantially in recent years, but treatment outcomes are still inadequate and the disease has a poor prognosis. Proteasome inhibitors and stem cell transplantation significantly improve patient survival; however, relapse over time and resistance to chemotherapeutic agents and molecular-targeted agents are problematic. Furthermore, aging of the population has led to an increase in the incidence of myeloma. MM remains challenging to cure, and even when treatment is successful, the disease is likely to gradually diminish in responsiveness to therapy, eventually resulting in relapse.
Adoptive transfer of genetically modified T cells to recognize malignancy-associated antigens is a promising approach for cancer therapy. 1 –4 For B-lineage malignancies, substantial progress has recently been made in developing adoptive T cell approaches using antiCD19 chimeric antigen receptors (CARs). 5 –9 However, CD19 is rarely expressed in malignant plasma cells. One antigen that has received considerable attention as a selective therapeutic candidate is BCMA. 10 As of December 2023, the U.S. Food and Drug Administration (FDA) has approved idecabtagene vicleucel and ciltacabtagene autoleucel as BCMA-targeted CAR-T cell therapies for relapsed or refractory MM.
MM is a hematopoietic malignancy characterized by monoclonal immunoglobulin (paraprotein) production and end-organ damage, including lytic lesions in bones, renal impairment, hypercalcemia, and anemia. 11 The mechanism underlying this disease involves the abnormal proliferation and accumulation of malignant plasma cells in the bone marrow. Despite the promising objective response rates of BCMA-CAR-T therapy, relapse in most patients and low level or loss of BCMA in a subset of tumor cells have been suggested as probable escape mechanisms. 12 –15 Although much attention has been paid to the future development of BCMA-CAR, the most reported benefits of BCMA-CAR have been discussed, but there have been no reports on the effects of CAR-T therapy on anemia and bone lesions in any animal model. We constructed a BCMA-CAR and verified its efficacy in the experiments presented here. Furthermore, the effectiveness of BCMA-CAR-T cell therapy on anemia and bone lesions associated with myeloma progression was confirmed in a mouse model.
MATERIALS AND METHODS
Cell culture
Cell lines and primary human peripheral blood mononuclear cells (PBMCs) used in this study were explained more extensively in the Supplementary Data.
Hybridoma generation for antihuman BCMA monoclonal antibody production
To obtain a monoclonal antibody against human BCMA, we immunized 6-week‐old BALB/c mice with recombinant human BCMA/TNF receptor superfamily member 17 (TNFRSF17) Fc chimera (Cat. No. 193-BC; R&D Systems). Immunization, fusion, screening, and cloning of hybridomas were performed by ITM Co., Ltd. (Matsumoto, Nagano). Six candidate hybridoma clones were selected for the study.
Identification and verification of hybridoma-derived monoclonal antibody variable region sequences
Total RNA was extracted, and amplification and analysis of cDNA generated from each hybridoma was performed using the SMARTer® RACE 5′/3′ kit from Clontech (Takara Bio, Shiga, Japan) and applied to isolation of antibody light and heavy chain variable gene sequences. Because all isotypes of antibodies produced from each hybridoma were IgG1 and kappa, PCR of the L-chain variable region was performed using a mixture of UPM sense primers corresponding to the 5′ end of the 5′-RACE cDNA and antisense primers corresponding to the end of the L-chain stationary region CL with primer sequence number 1 (Table 1). PCR of the H-chain variable region was performed using a mixture of UPM sense primers corresponding to the 5′ end of the 5′-RACE cDNA and antisense primer corresponding to the end of the H-chain stationary region CH1 with primer sequence number 2 (Table 1). Next, the PCR products were subjected to an infusion reaction using a cloning vector (Takara Bio Inc.). Ten colonies obtained from each DNA sample encoding the light chain variable region (VL) and heavy chain variable region (VH) regions were selected and cultured. Each plasmid was sequenced by Eurofins Genomics K.K. (Tokyo, Japan). The obtained sequences were searched using basic local alignment search tool (BLAST), and all clones showed homology to the IgG L- or H-chain sequences, confirming that the cloned PCR products were DNA fragments containing nucleic acids encoding the variable regions of the L- and H-chains. The amino acid sequence was deduced by searching for the start codon ATG, which is located upstream of the 5′ end of the stationary region and is in frame with the stationary region. The estimated amino acid sequences of each clone’s light and heavy chains were compared using ClustalW2 Multiple Sequence Alignment to maximize homology and then compared for complementarity-determining regions, as defined by the Kabat method.
Primer Sequences Used in This Study. N Indicates A, C, G, or T. The Letter “N” in Primer Sequences Depends on the DNA Sequences of the Antibody of Interest
CAR construction
The retroviral vector pMEI-5, containing only the long terminal repeat and packaging signal (Ψ sequence) of the moloney murine leukemia virus (MoMLV) genome but not the gag, pol, and env coding sequences (Takara Bio Inc.), was used as the backbone for all CAR constructs. The L- and H-chains of each antibody gene were amplified by PCR and subcloned by the infusion method into the pEX-A2J2-1G/18 plasmid, which was double-digested with SmaI and SfoI to construct scFv constructs with a linker sequence of 18 aa. Specifically, to generate the L18H construct for each antibody gene, the L-chain was amplified with primers 3 and 4 (Table 1), and the H-chain was amplified with primers 5 and 6 (Table 1). To generate the H18L construct for each antibody gene, the H-chain was amplified with primers 7 and 8(1), and the L-chain was amplified with primers 9 and 10 (Table 1). The L15H construct was created by PCR amplification using pShuttle-L18H as a template with primers 11 and 12 (Table 1) for mutagenesis using the infusion method. Similarly, the L20H construct was built by PCR amplification using pShuttle-L18H as a template with primers 13 and 14 (Table 1). The pShuttle plasmid was double-digested with PmlI and NotI to cut out and purify the scFv fragments, and then subcloned into pMEI-5/28z with pre-subcloned hinge, transmembrane, and intracellular regions of CD28 and the intracellular region of CD3ζ to construct BCMA-CAR expression plasmids with each scFv sequence.
Retrovirus production
293T cells were transfected with pMEI-5 retroviral vector plasmid and retroviral packaging plasmid (gag-pol expression plasmid pGP and VSV-G pseudotyped envelope expression plasmid pVSV-G) using the calcium phosphate method. The viral supernatant was harvested at 48 h after transduction. A quarter volume of the Retro-X concentrator was added to the collected culture supernatant, mixed well, and allowed to stand at 4°C for 24 h. After centrifugation at 1,500× g for 45 min at 4°C, the supernatant was removed, and the pellet was resuspended in 2 mL medium. PG13 cells were exposed to the concentrated viral supernatant in the presence of 8 μg/mL polybrene and incubated overnight at 37°C in 5% CO2. Single-cell clones were isolated using the limiting dilution method and cultured for 7–10 days in an incubator at 37°C in 5% CO2. Cells were incubated for 48 h in an incubator set at 32°C and 5% CO2. Cell debris was removed by filtration through a 45-µm filter, and the purified viral supernatant was aliquoted and stored at −80°C. The retroviral RNA titer was measured using the Retrovirus Titer Set for Real-Time PCR Kit (Takara Bio Inc.) according to the manufacturer’s instructions.
T cell transduction
For activation of PBMCs, 5 µg/mL CD3 monoclonal antibody (OKT3) (Thermo Fisher Scientific) diluted with ACD-A solution (Thermo Fisher Scientific) and 20 µg/mL RetroNectin (Takara Bio Inc.) were added to six-well plates. PBMCs were seeded onto OKT3/RetroNectin-coated plates for 4 days to promote the activation and proliferation of T cells. Retrovirus vector solution (diluted to 4.0 × 109 copies/mL with PBS) was added to RetroNectin-coated plates. The plate was centrifuged at 32°C for 2 h at 1,960×g to promote the adsorption of virus particles on the surface of the RetroNectin-coated plates. The virus diluent was removed from the plates and washed with 2 mL PBS supplemented with 1% BSA, and OKT3/RetroNectin-activated T cells (diluted to 4.0 × 105 cells/mL in medium) were added to the virus-adsorbed plate. The plates were incubated overnight at 37°C in 5% CO2 for transduction. The next day, transduced T cells were collected and expanded for 10 days using a scale-up culture method. The use of clinical samples, including PBMCs, was reviewed and approved by the Ethical Board of Jichi Medical University, and written informed consent was obtained from all donors.
In vitro reporter assay
Jurkat-1928z-iReporter cells or gene modified T cells (GMCs) were cocultured with target tumor cells for the times indicated in the figure legends. Luciferase activity was measured using the Bright-Glo Luciferase Assay System (Promega, Madison, WI, USA) with a Fluoroscan.
CAR-T cell cytotoxicity
To monitor cytolytic activity, increasing numbers of CAR-T cells were cocultured with tumor cells labeled with calcein-AM in 96-well plates. This is further described in the Supplementary Data.
Flow cytometry
Antibodies used in this study were explained more extensively in the Supplementary Data.
Enzyme-linked immunosorbent assay
interferon gamma (IFN-γ) production was measured using a Human IFN-γ enzyme-linked immunosorbent assay (ELISA) Kit (Thermo Fisher Scientific Inc.). Serum λ free light chain (FLC) concentrations were measured by ELISA [IgG λ (f & b], Human, ELISA Quantitation Kit; BETHYL laboratories, Inc., E80-116) (Supplementary Data).
Adoptive CAR-T cell transfer in tumor-bearing mice
We used 6- to 8-week-old male NOG (NOD/SCID/IL-2Rγnull) mice (Central Institute for Experimental Animals [Tokyo, Japan]) under a protocol approved by the Jichi Medical University Animal Care and Use Committee. Mice were inoculated with 1 × 106 ELuc-U266 cells by intracardiac chamber injection, followed by infusion of a net amount of 1 × 106 CAR+ T cells 42 days later. Bioluminescence imaging was performed using an IVIS Imaging System (PerkinElmer) with Living Image software (PerkinElmer) to acquire imaging datasets.
Immunohistochemistry
At 21 days after therapy, the femurs were removed from the mice, and paraffin-embedded tissues were prepared. Immunoreactivity for IgE was visualized with 3,3-diaminobenzidine using a peroxidase-based Histofine Simple Stain Kit (MAX PO R, Nichirei). This is further described in the Supplementary Data.
Sample preparation for hematological analysis and μCT scanning and 3D reconstruction of mice femurs
Blood was collected from mice before and 21 days after therapy. The hematological status of the mice was assessed by Oriental Yeast Co., Ltd., Nagahama LSL (Nagahama, Japan). A portion of the collected blood was transferred to blood-separ tubes (No. 31203; Immuno-Biological Laboratories) for FLC measurements. At 21 days after therapy, spleens were removed from the mice and their weights were measured. At 21 days after therapy, femurs were removed from mice and µCT scanning and 3D reconstruction of mouse femurs were performed by the Kureha Special Laboratory Co., Ltd. (Iwaki, Fukushima).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9 software (GraphPad Software, MA, USA). No statistical method was used to determine the sample size. Statistically significant differences between two groups were assessed using a two-tailed paired or unpaired t-test. Comparisons between more than two groups were performed using analysis of variance with Tukey’s multiple comparison test. In the mouse experiments, the overall survival of the mice was depicted by a Kaplan–Meier curve, and the survival difference between the groups was compared using the log-rank test. Statistical significance was set at p < 0.05. The statistical tests used for each figure are described in the corresponding figure legends. No statistical method was used to determine the sample size.
RESULTS
Generation of antihuman BCMA mABs and BCMA-CARs
Three BALB/c mice were immunized with recombinant human BCMA using the iliac lymph node method. Iliac lymph nodes were harvested from immunized mice and fused with lymphocytes and SP2/0 myeloma cells. Finally, we cloned six antiBCMA mAb-producing hybridomas. The antibody isotype produced by each hybridoma is IgG1κ. Antibody specificity was assessed by comparing the binding signals in cells expressing BCMA to those in control cells. We used K562-BCMA, U266, and RPMI8226 cells expressing BCMA, and K562 and K562-CD19 cells as control cells (Supplementary Fig. S1). A commercially available antiBCMA antibody (clone 19F2; BioLegend) was used to bind BCMA on the surface of cells (Supplementary Fig. S2). We analyzed the DNA sequences encoding VL and VH, and determined the CDR1, CDR2, and CDR3 of VL and VH. Based on a series of analyses, we inferred that the antibodies produced by each hybridoma clone were different. CARs containing the antigen-binding fragments of each antiBCMA antibody were prepared starting from the N-terminus, and comprised the signal peptide sequence of the granulocyte macrophage colony stimulating factor (GM-CSF) receptor, the VL region of the antiBCMA antibody, a linker sequence (18 aa), the VH region of the antiBCMA antibody, the hinge and transmembrane regions of the CD28 molecule, the costimulatory signaling moiety of the CD28 molecule, and the signaling domains of the CD3ζ molecule. This is referred to as L18H-CAR. Nucleic acids encoding CARs, in which the order of the VL and VH regions was swapped, were also generated. This is referred to as H18L-CAR. The structure of the CARs is shown in Figure 1A.
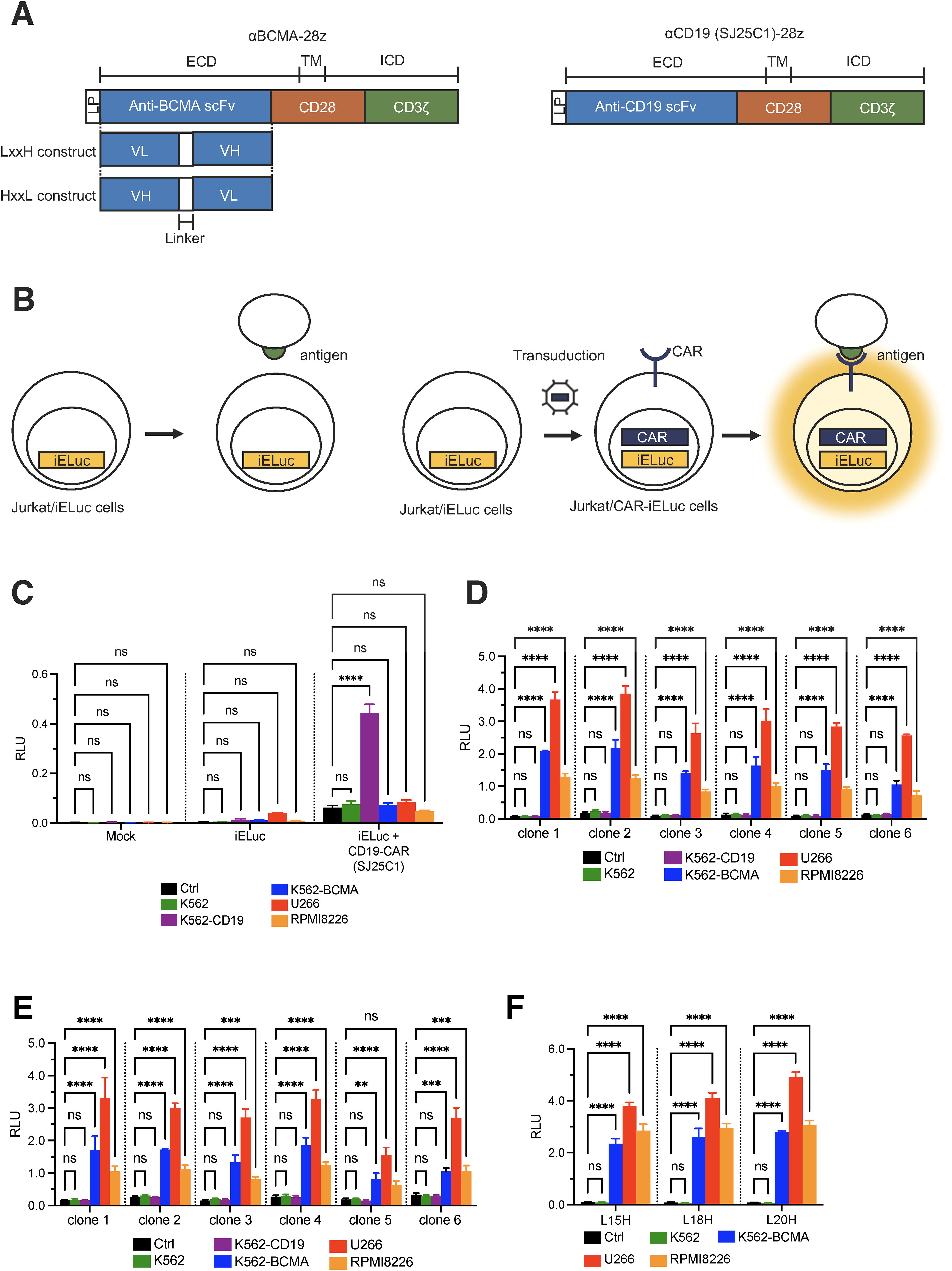
Generation of BCMA-CAR.
Previously, we constructed self-inactivating (SIN) retroviral vectors containing four or six NFAT-REs, followed by a minimal IL2 promoter and luciferase gene. First, we generated Jurkat cells transduced with this expression system (Jurkat/iELuc). Because luciferase expression is induced in CAR-transduced Jurkat/iELuc cells, these transduced cells can induce reporter gene expression when cocultured with target antigen-positive cells (Fig. 1B). The specificity and reactivity of CARs can be compared and evaluated based on luminescence intensity. Preconfirmation using CD19-CAR (scFv derived from SJ25C1 clone) established that luciferase luminescence was measured only in the reaction with CD19-positive K562-CD19 cells (Fig. 1C). Jurkat/iELuc cells transduced with BCMA-CAR were cocultured with the target cells. After 18 h, the luminescence intensity of the transfected cells was measured. In a study in which LH constructs were transduced into Jurkat/iELuc cells, luciferase luminescence was measured in all CARs under coculture conditions with BCMA-positive target cells. In particular, CARs derived from clone 1 showed the highest reactivity, with a 36-fold higher luminescence intensity when cocultured with U266 cells compared with the control (Fig. 1D). However, in a study in which HL constructs were used, none of the CARs showed luminescence intensities exceeding that of clone 1 from the L18H relative to the control because the luminescence intensity ratio of the control was higher than that of LH (Fig. 1E). The order in which the light and heavy chains are linked causes differences in reactivity with the target cell and tonic signaling at the basal level.
Next, we tested how the CAR reactivity changed when the linker length was shortened (15 aa) or lengthened (20 aa). CAR derived from clone 1 with linker lengths of 15 aa or 20 aa was designated BCMA-CAR (L15H and L20H, respectively). Interestingly, the linker length also causes a difference in basal-level tonic signaling. In BCMA-CAR T cells, the long linker showed lower tonic signaling and better reactivity with BCMA-positive cells (Fig. 1F). Under the coculture conditions of L20H-CAR and U266 cells, the luminescence intensity was 62.8-fold higher than that of the control.
Therapeutic efficacy of BCMA-CAR-T cells
Retroviral vectors encoding CARs (L20H) were used to transduce human PBMCs. After transduction, we observed high levels of cell surface expression of BCMA-CAR in GMCs (Fig. 2A–C). GMCs showed redirected cytolysis toward BCMA-positive BCMA-K562 and U266 cells, but not toward BCMA-negative K562 cells (Fig. 2D), and produced large amounts of IFN-γ when cultured overnight with the BCMA-expressing target cell lines BCMA-K562 and U266 (Fig. 2E).
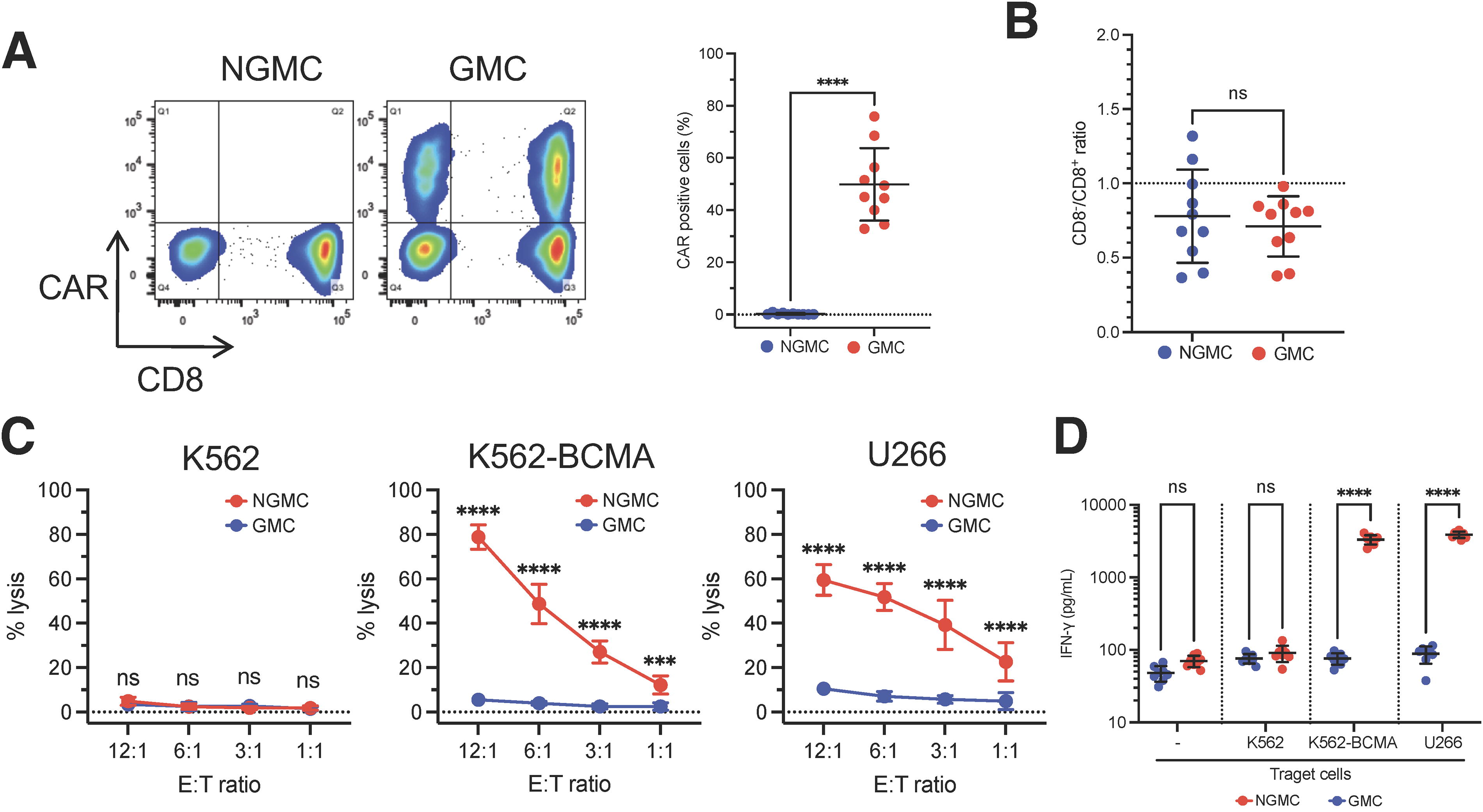
In vitro efficacy of BCMA-CAR T-cells. (A) Transduction efficiency of BCMA-CAR was measured by flow cytometry (n = 10 different donor samples).
We established tumor mouse models by injecting ELuc-expressing U266 cells via the left ventricular cavity into NOG mice. Generally, mice were irradiated prior to transplantation to enhance tumor cell engraftment, but we allowed systemic progression of MM in the bone marrow over 42 days without prior irradiation (Supplementary. Fig. S3A–C). U266 cells did not infiltrate multiple organs such as the liver, spleen, and lungs of NOG mice, which reflects the clinical manifestations observed in myeloma patients. Serum free light chain was detected in plasma (Supplementary. Fig. S3D). In our mouse model, hind limb paralysis appeared 10–11 weeks after inoculation with U266 cells. According to previous reports, the injection of U266 cells after 2.4 Gy irradiation resulted in hind limb paralysis after 6 weeks. 16 The absence of irradiation reproduces the phenomenon in which myeloma cells generally increase slowly and are characterized by a slow onset of symptoms.
All mice treated with \GMCs achieved a complete response with dramatic regression of all tumors occurring between days 6 and 15 after GMC infusion (Fig. 3A–C). In contrast, the tumors continued to increase in size in all mice that received saline or nongene modified T cells (NGMCs). Although transient weight loss was observed in mice that received GMCs, the body weight gradually recovered after the tumor disappeared, and the mice showed no signs of toxicity, including xenogenic graft versus host disease (GVHD), during this experiment (Fig. 3E). All the mice treated with GMCs survived the observation period (Fig. 3F). Histological analysis of the bone marrow showed that, despite the increase in MM in the bone marrow of mice treated with saline or NGMCs, no MM was detected in the bone marrow of GMC-treated mice, indicating a histology similar to that of normal mice (Fig. 3G). In addition, λFLC was not detected in the sera of mice that received GMC infusions (Fig. 3H).
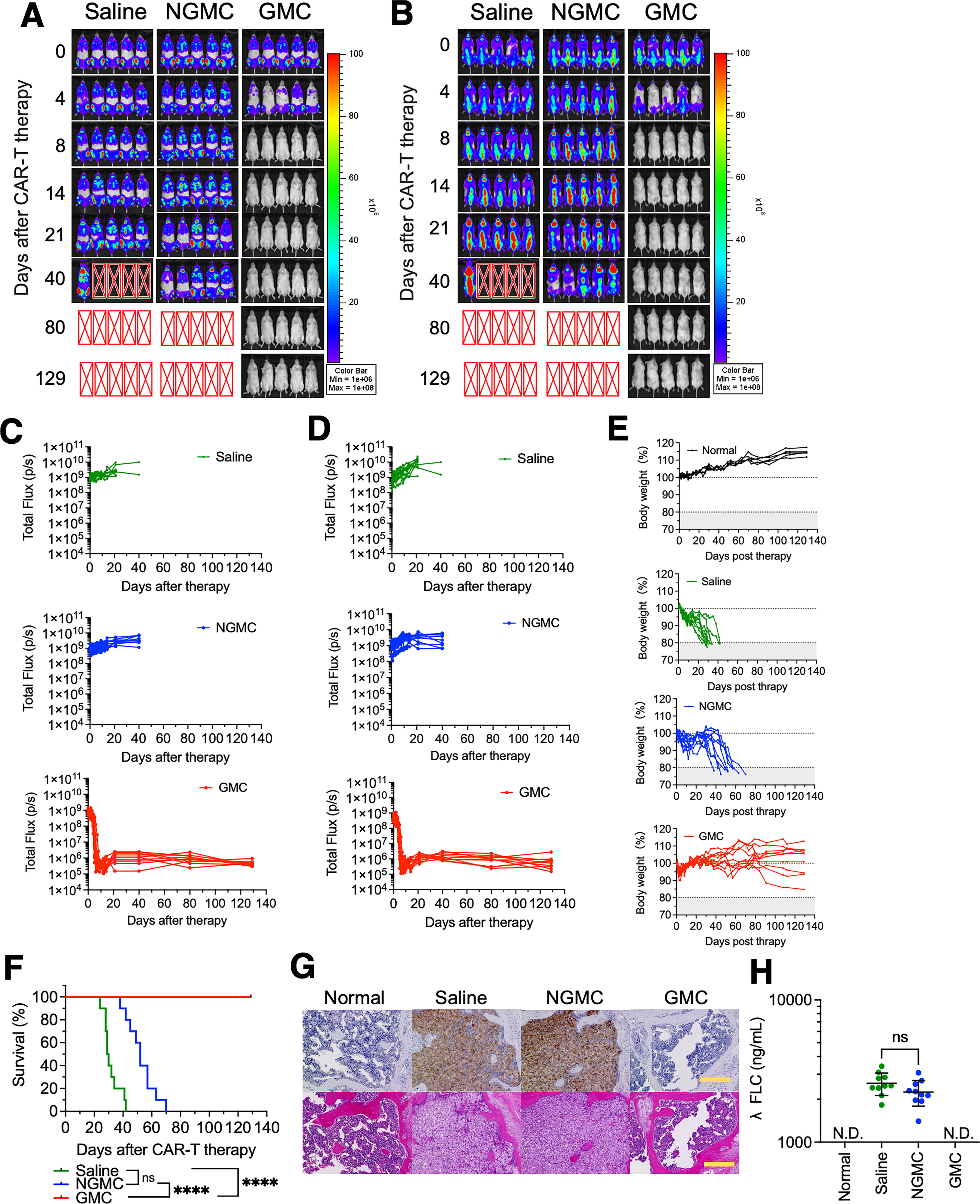
In vivo efficacy of BCMA-CAR T-cells.
An inducible gene expression system was used to analyze whether infused CAR-T cells showed tumor-specific responses 17 (Fig. 4A and Supplementary Fig. S4). In the iELuc-T cell-treated group, partial infused cell activation was observed, but tumor cells continued to grow thereafter (Fig. 4B, C, F, and G). In contrast, in the CAR/iELuc-T cell-treated group, systemic activation of infused cells was observed, and the tumor cells disappeared (Fig. 4D, E, H, and 1). Thus, the infused CAR-T cells exhibited tumor cell-specific cytotoxic responses. Interestingly, the activation of infused T cells also reduced rapidly with the disappearance of the tumor cells. To verify the persistence of CAR-T cells produced in this study in mice, mononuclear cells were collected from the peripheral blood of mice 80 days after CAR-T cell injection to measure provirus counts, but the provirus was not detected (data not shown). However, the vector structure used in this study was similar to that of conventional BCMA-CAR, and the expected persistence of CAR-T cells produced in this study in mice should be comparable.
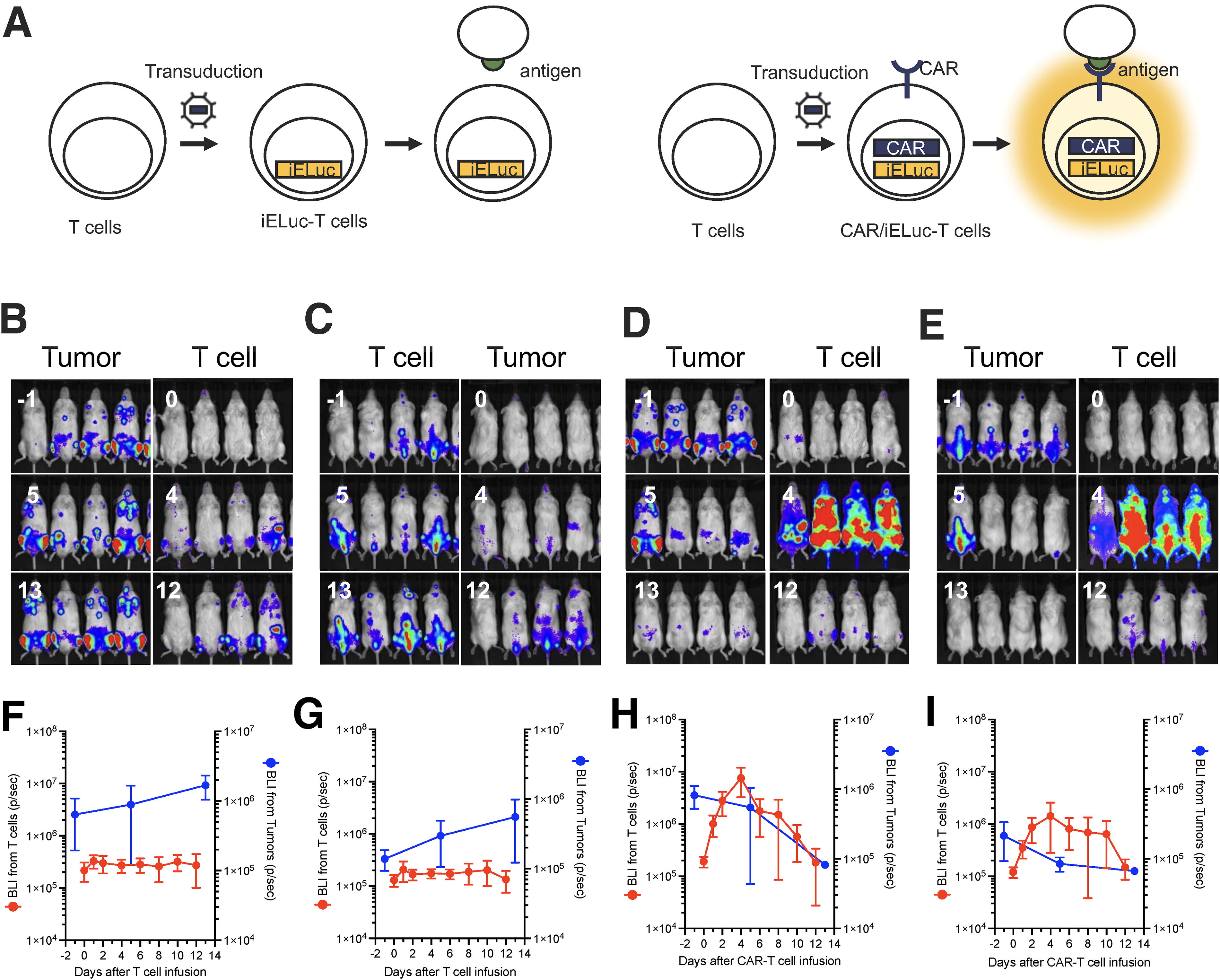
Target-specific activation of infused BCMA-CAR T-cells.
Inhibitory effect on the progression of anemia
Myeloma cells infiltrating the bone marrow impair the function and structure of erythroblastic islands by secreting cytokines. It was found that the spleen weight was markedly increased in the tumor-bearing mice compared with normal mice (Fig. 5A). This was assumed to be due to the infiltration of U266 cells into the bone marrow, which inhibited normal intramedullary hematopoiesis, resulting in enhanced extramedullary hematopoiesis in the spleen. Spleen weight was also decreased in the GMC-treated group, although it was higher than normal, indicating a return to normal weight. Moreover, fetal liver hematopoiesis in mice disappears approximately 2 weeks after birth; however, under acute experimental anemia, extramedullary hematopoiesis occurs in the livers of adult mice. 18 Therefore, it is presumed that it is technically difficult to reproduce the anemia pathology of MM patients in mice. In this study, MCH was significantly decreased in tumor-bearing mice at the beginning of treatment, but the RBC and reticulocyte counts tended to increase. However, mice treated with saline and NGMCs showed a tendency toward anemia over time, and RBCs and reticulocytes decreased. In contrast, in mice treated with GMCs, the progression of anemia was prevented and resembled that observed in the normal mice (Fig. 5B).
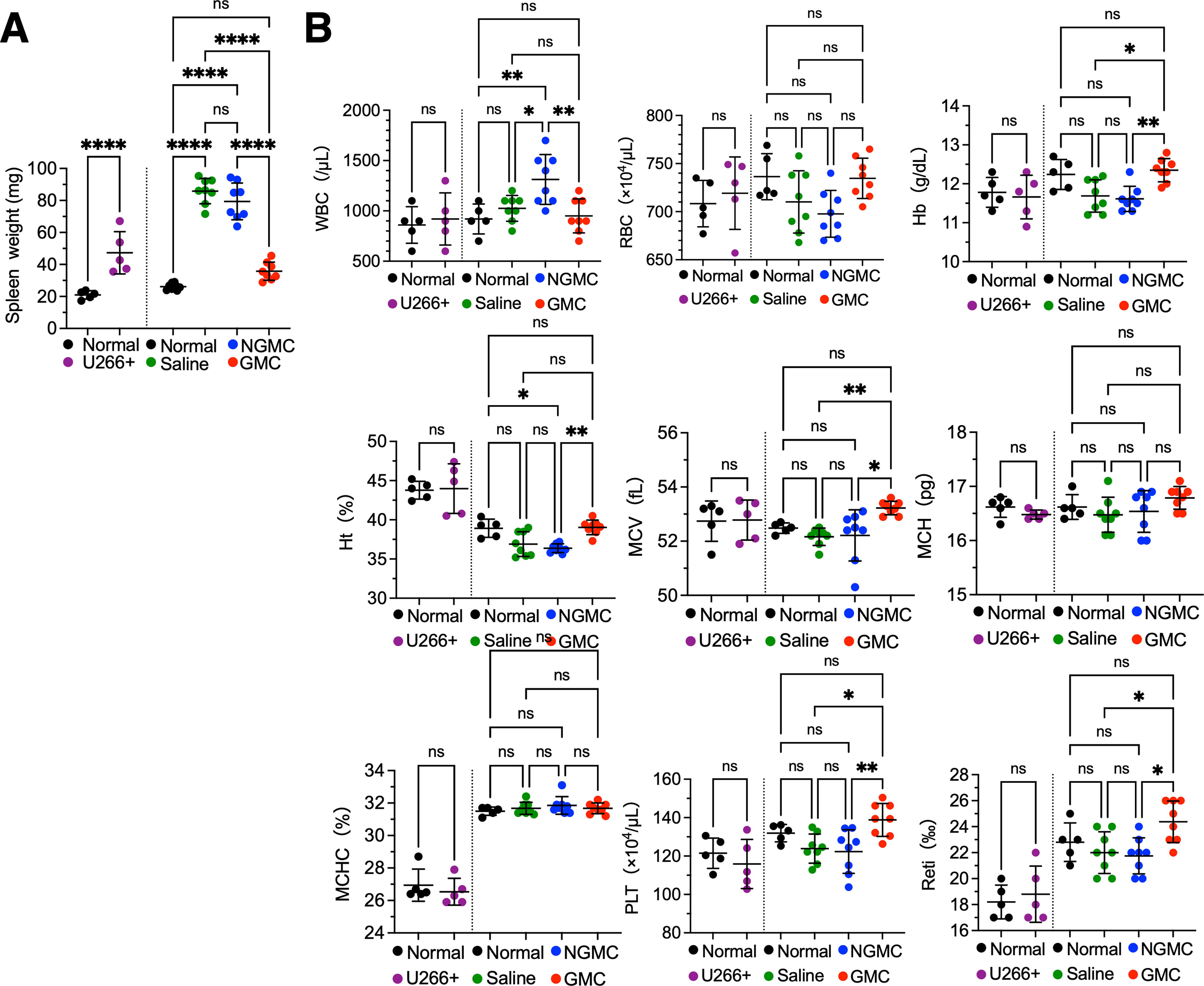
Hematological parameters.
Inhibitory effect of CAR-T therapy on the progression of bone lesions
In myeloma patients, tumor cells stimulate osteoclasts (OCs), causing an imbalance between them and leading to increased destruction and consequent weakening and brittleness of the bones. As a result, the spinal cord is compressed by the myeloma mass that has destroyed the bone and popped out or by the vertebrae deformed by the fracture, causing pain, muscle weakness, numbness, paralysis in the limbs, and significantly reduced quality of life. Trabecular bone morphology was assessed using microCT. At the start of treatment, tumor-bearing mice showed slightly advanced bone lesions compared with normal mice. However, the bone surface was similar to that in normal mice (Fig. 6A, C). Mice treated with saline or NGMCs showed progressive bone lesions. Severe destruction of the condyle and the bone surface was observed (Fig. 6B, C). In contrast, in mice treated with GMCs, the progression of bone lesions was strongly prevented, and the bone surface was similar to that of normal mice. Although the phenomenon observed in this study appears to have been the inhibition of bone lesion progression with GMC administration, further studies are required to elucidate the long-term effects of CAR-T therapy on bone regeneration.

3D microCT images of mouse femurs.
DISCUSSION
In this study, we focused on the effect of CAR-T cells on anemia and bone lesions associated with myeloma exacerbation, although the efficacy of BCMA-CAR has previously been demonstrated. To date, therapeutic evaluation approaches for CAR-T therapy for myeloma have been limited to imaging systems, and there have been no reports on the assessment of laboratory values and concomitant conditions that are routinely used in clinical practice in mouse models. This study revealed that evaluation methods such as 3D-CT and FLC could function in a mouse model of CAR-T therapy and demonstrated the efficacy of CAR-T using these modalities.
Myeloma bone disease (MBD) is a severe complication of MM that severely hinders activities of daily living and negatively affects patient survival. Osteocytes and their surrounding microenvironment play crucial roles in MBD development. Osteocytes derived from osteoblasts (OBs) can contribute to OC activation by releasing several cytokines including osteoprotegerin, receptor activator of NF-κB ligand (RANKL), Dickkopf-1 (DKK-1), and others. 19 In normal bone remodeling, OCs remove damaged bone tissue, and OBs form new bone tissue, maintaining a balance between the two processes. However, this balance is disturbed in MM patients. The production of DKK-1 been attributed to OBs, BMSCs, and myeloma cells. DKK-1 impedes the signaling pathway by competitively binding to the receptors of the Wnt pathway, thereby stimulating OC activity and hindering new bone formation. 20 –22 Among patients recently diagnosed with MM, those with bone lesions visible via imaging had significantly higher levels of DKK-1 expression at both the genetic and protein level than those without bone lesions. Because elevated DKK-1 levels impede OB differentiation, 23 DKK-1 antibodies enhance bone density and decrease osteolytic lesions in myeloma-bearing mice. 24,25 Our results showed that even treatment with BCMA-CAR could inhibit the progression of bone lesions by eliminating myeloma cells but did not lead to recovery due to short-term observations. Further long-term observations are required to assess the recovery of the damaged trabecular network.
The rapid development of BCMA-targeting CAR-T cells in MM since their first administration in 2014 to the FDA approval of ide-cel in 2021 holds great promise for the future of MM treatment. However, some studies have shown that relapse after CAR-T cell therapy is caused by a loss of CAR-T cell persistence in the body. To overcome these limitations and achieve therapeutic success, current clinical trials are evaluating whether earlier lines of CAR-T therapy can lead to long-term remission. Many different approaches have been investigated to improve CAR-T cell therapy through the development of next-generation CARs. As demonstrated in our study, therapies targeting myeloma cells are not sufficient to inhibit the progression of bone lesions; therefore, it is critical to develop novel supportive therapies and to assess bone lesions appropriately in the process of developing such therapies. CAR-T cells to produce antiDKK-1 antibodies may be a promising strategy to kill myeloma cells and ameliorate MBD in parallel. Finally, although this study represents the therapeutic effects of BCMA-CAR-T cell therapy, such as inhibition of progression or improvement of complications associated with cancer progression, better outcomes may be demonstrated in the future for enhancing CAR-T cell function against MBD.
Footnotes
ACKNOWLEDGMENTS
AUTHORS’ CONTRIBUTIONS
Conceptualization, methodology, investigation, writing—original draft, and visualization were performed by R.U. Investigation, writing (review and editing), and visualization were performed by K.Oh. and T.T. Supervision and writing (review and editing) were undertaken by J.M. and K.Oz.
AUTHOR DISCLOSURE STATEMENT
T.T. and J.M. are employees at Takara Bio. No other disclosures are reported.
FUNDING INFORMATION
Funding for this study was provided by Takara Bio Inc. and was supported by JSPS KAKENHI (Grant numbers JP24700995 and JP16K18459).
SUPPLEMENTARY MATERIAL
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.