
Abstract
The study of β-hemoglobinopathies and associated β-globin genes has revealed that genetic elements, such as the Locus Control Region (LCR) or the replication Initiation Region (IR) of the β-globin gene locus, are essential for the regulation of β-globin genes replication and expression. The LCR at 5′ of the β-globin genes plays major role in the intricate regulation of transcription of the “β-like globin genes” expression in situ and in gene therapy protocols by viral gene transfer, ensuring globin gene expression independent from integration site and exerting a critical role in chromatin organization and boundary formation. The IR element, located at the 5′ site of the HBB gene promoter, functions as the initiation point for physiological, bidirectional DNA replication, both in situ and within an episomal vector, and induces replication in positions that do not possess such capacity. It enhances plasmid replication, establishment, and transgene expression in the descendants of transfected human CD34+ cells during colony-forming cell assays. A third required genetic element is the promoter of the transgene(s). This is either the HBB gene native promoter or the CD34+ cell-functional ubiquitous promoter spleen focus-forming virus. Both promoters, in in vitro studies, can direct accurate, efficient transcription from episomal, S/MAR-based vectors. Mutations in the HBB gene native promoter as well as in LCR and IR lead to β-thalassemia. Another genetic element, the S/MAR, deriving from the 5′ of the human β-interferon gene, ensures plasmid nonintegration and long-term nuclear retention in the prototype episomal vector pEPI-1 and derivative episomal vectors. Such S/MAR-based episomal vectors form the basis from which the genetic elements collectively— HBB gene promoter, LCR, and IR—represent a comprehensive model for the design of efficient episomal vectors with efficient transcription, replication, and long-term nuclear retention of vector for gene therapy applications for the β-hemoglobinopathies within the context of gene addition strategy.
Keywords
INTRODUCTION
Nonviral episomal vectors are under development as a valid alternative to viral vectors, presenting a safer profile, as nonintegrating by design, and thus circumventing the problems of insertional mutagenesis/oncogenesis. The development of episomal vectors, carrying the physiological HBB gene for gene therapy of β-hemoglobinopathies, began 22 years ago with the study of a 38-kb beta-Locus Control Region (LCR) minilocus replicating episomal vector. In that study, the human beta-globin LCR (beta-LCR) was used as a model to show that such control elements of gene expression are capable of driving high levels of tissue-specific transgene expression in cultured cells that are stably transfected with a replicating episomal vector. 1
We mark out the study of the β-hemoglobinopathies and the respective genetic, regulatory elements within the β-globin gene locus as a representative model for the formulation of an effective, noninvasive genome and low-cost episomal gene transfer system for the gene therapy applications of these diseases.
The β-hemoglobinopathies, historically, have been the subject of extensive and in-depth studies on globin genes, due to the accessibility of blood cells, but also to the informative complexity of their genomic organization. Molecular analysis of the structure and expression of the β-globin genes, both in physiological and in β-hemoglobinopathy conditions, led to the unraveling of the complexity of the transcriptional regulation of the β-globin genes’ expression. Thus, a deeper understanding of transcription as the critical stage of a gene’s expression generated a model for the medical, molecular analysis of human and hereditary diseases, as well as a paradigm for the molecular analysis of genes within the field of human genetics.
The β-globin genes form a cluster located within the respective genetic locus in chromosome 11p15.4. 2 Impairment of their expression is the basis of the β-hemoglobinopathies—sickle cell diseases (SCD) and β-thalassemia. These diseases, which have very little choice of conventional therapy in most cases, are caused by 1 of ∼350-point mutations that occur mainly within the β-globin gene. They are monogenic, hereditary blood disorders, the majority of which follow the Mendelian recessive mode of inheritance. Dominantly inherited β-thalassemia also exist due to mutations that cause functional deficiency of the β-globin gene followed by precipitation of the β-chains and are associated with hematological features that are typical of β-thalassemia. 3 Another class of mutations causing β-thalassemia are numerous deletions, ranging from a few hundred base pairs to just over 500 kilobase pairs, some of them removing part or the whole of the HBB gene and some leaving intact the “β-like globin genes” of the cluster and removing the regulatory sequences at the 5′ site of the genetic locus. β-Thalassemia, deriving from decreased β-globin (β+) peptide chains or absent β-globin (βo) peptide chains, covers a number of hematopoietic disorders with similar molecular bases, the so-called β-thalassemic syndromes. 2
Compilations of all published mutations worldwide, as well as information on the β-globin gene variations affecting hemoglobin disorders, are found in Web Databases, such as the old Globin Gene Server (https://globin.bx.psu.edu/) and the more recent Interactive Database of Hemoglobin Variations, the IthaGenes (https://www.ithanet.eu/db/ithagenes).
The great genetic heterogeneity of the disease generates a wide spectrum of diverse β-thalassemias, the most prevalent of the rare diseases 2 that are manifested as an effect on the β-globin protein produced. Specifically, they may exert an effect on the structure of the protein, and hence its function, with prominent cases like SCD, or they may reduce the level of β-globin mRNA produced, even abolishing it altogether, giving rise to β-thalassemias. Additionally, a number of modifiers of the globin genes expression exist, which contribute to the fine-tuning of the expression program of the β-globin genes regarding tissue and developmental stage specificity. 4
This review focuses on the lessons learned from the study of hemoglobinopathies as disease states, often associated with the absence or the attenuation of crucial regulatory elements or with point mutations that lower the expression of the HBB gene. We will explore how loss of function of elements, such as a cell-specific/cell-functional promoter, the β-globin LCR, or the β-globin replication Initiation Region (IR or the β-globin Replicator), relate to hemoglobinopathies, revealing the function of these regulatory elements and thus, presenting a model for the design and development of episomal, S/MAR-based vectors for the genetic therapy of these diseases. An extensive presentation of these elements is included in the review by Mulia et al. 5
GENE THERAPY FOR THE β-HEMOGLOBINOPATHIES
The β-hemoglobinopathies were among the first diseases to be considered for gene therapy, in which the patients’ own stem cells are modified to carry the physiological HBB gene with its regulatory elements, according to the “gene addition” strategy. After a long route of development, originally by applying retroviral vectors and later lentiviral vectors, a landmark in the gene therapy for β-hemoglobinopathies was the cure of β-thalassemic mice by the use of lentivirus-encoded human β-globin. 6 Self-inactivating (SIN) viral vectors greatly increased the safety of viral gene transfer, and therefore, the number of clinical trials with patients with β-thalassemia, culminating in the formulation of the therapeutic product, Zynteglo. This therapeutic agent was provided authorization for marketing in Europe by the European Medicines Agency (EMA), but later was withdrawn after a reimbursement dispute (https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo). A therapeutic product, based on the same scientific context, was subsequently authorized by the Food and Drug Administration (FDA), USA.
Following many years of studies, clinical trials, and validation processes, the β-hemoglobinopathies were recently added to the list of diseases for which gene therapy is a therapeutic option. This is, so far, a safer and more potent therapeutic gene therapy strategy, as a one-time curative, autologous treatment, based on a self-inactivating lentiviral system for the gene transfer of the physiological HBB gene, leading to a great success, celebrated as such. 7 The current strategy of lentiviral gene transfer to treat β-hemoglobinopathies has its limitations too, in that it provides cure only for the transfusion-dependent β+/β+ patients and not for most β0/β0 patients, who are absolutely deficient in β-globin chains production. Furthermore, a number of challenges still remain that need to be addressed. A few, albeit important, data obtained from monitoring transduced patients raised concerns. A clinical trial with thalassemic patients reported (moderate) clonal expansions associated with vector integrations near genes related to cancer. 8 In another clinical trial, with SCD participants, acute myeloid leukemia/myelodysplastic syndrome was developed, possibly due to insertional mutagenesis, as the transgene was present in the malignant cells. 9 As an expression of the globin β-transgene is normally erythroid-specific, these data raised concerns about the possible nonerythroid function of these lentiviral-based vectors. Indeed, it has been demonstrated that the lentivirally encoded HS1 and HS2 parts of LCR can be activated not only in hematopoietic stem cells but also in myeloid progenitor cells, resulting in the activation of the transgene, although the addition of an insulator can lessen this effect. 10
A number of other causes of concern exist. There is the risk of secondary hematological malignancies generated from the action of factors deriving from the disease, namely, the treatment and the circulating cancer cells. 11 There is also the immunological response to proteins encoded by the vector, which are not manifested so far by lentiviral vectors but, apparently, are not excluded. Another stabling block has been the complexity of the production of the therapeutic viral system to be delivered. Adding the high cost of the therapy along with the difficulty of its distribution and use in less developed parts of the World results in restriction of the broad use of this gene therapy strategy. It seems, therefore, that hematopoietic stem cell transplantation, when the need for a matching donor is met, remains the only cure for patients with transfusion-dependent thalassemia until gene therapy strategies are further improved and safe.
In parallel, other approaches are under development and undergoing clinical trials; in particular, the CRISPR-Cas gene editing, with off-target events under control, has been applied for hemoglobinopathies, 12 focusing on the correction of the mutated HBB gene in vivo, to restore the normal state of the β-globin chains production. The application of single-base editor, combining precision editing of high-efficiency and higher safety profiles than the double-strand-break-based approaches, through the use of DNA nickase, has been shown to be effective for the correction of the severe β-thalassemia mutation IVSI-110 in hematopoietic stem and progenitor cells. 13 This approach has not, as yet, produced a cure for β0/β0 transcription-dependent patient with thalassemia. Nevertheless, another strategy of utilizing CRISPR/Cas9 technology, the one of activating the γ-globin gene with the aim of generating a Hereditary Persistence of Fetal Hemoglobin (HPFH) condition, was successful, as significant amount of fetal hemoglobin continues to be produced in the adult individual, complementing for the lack of normal β-globin chains. This was achieved by the use of Exagamglogene autotemcel (Casgevy™), 14 which received approvals from the UK, the European Medicines Agency (EMA), and the American FDA agencies, as the first CRISPR/Cas9 genome editing therapy for the treatment of transfusion-dependent β-thalassemia and SCD.
The issue of finding a gene therapy option for curing patients with β-thalassemia is partially solved, with β-thalassemia on the whole presenting a complex and difficult disease entity, and a single strategy of treatment is unlikely to work for every patient with β-thalassemia. Therefore, the development of diverse therapeutic approaches is highly desirable.
THE EPISOMAL, S/MAR-BASED VECTORS FOR GENE THERAPY
Episomal, S/MAR-based vectors under development for gene therapy applications derive from eukaryotic episomes of plasmid origin, as opposed to viral origin. Episomes hosted by eukaryotes are extrachromosomal, circular DNA molecules that divide along with the chromosomes during mitosis and are bound to nuclear matrix by a “piggy-back” type of mechanism. Nuclear matrix is a largely proteinous, structural filamentous network within the compartments of nuclear architecture, and episomal vectors are attached through binding protein SAF-A (and possibly other proteins).15,16 Episomal vectors are of greater size than conventional plasmids—usually >5 kb—and, upon entering the nucleus of a recipient cell, they land on the nuclear matrix. Mitotic stability provided by S/MAR leads to the capacity of the episomal vectors to replicate and, importantly, to distribute the replicated plasmids equally to duplicated daughter cells during the successive mitotic divisions.17,18 Otherwise, episomes survive only transiently in the nucleus, as successive mitotic cell divisions of the host lead to a gradual plasmid loss. Nevertheless, episomal vectors within the cell nucleus recruit histones and can assemble nucleosomes by a process that is more organized for replicating episomes, forming minichromosomes, thus avoiding nuclease action and securing a longer nuclear retention. 19
Such episomal vectors usually stay in the nucleus as open-access episomes, but they may also integrate into the endogenous DNA of the cell. Integration of episomal vectors is a rare event involving accidentally linearized vector molecules, which can be inserted into chromosomal DNA breaks through canonical, nonhomologous end joining, independently of DNA sequence complementarity. Intact episomes may be integrated by microhomology end joining during their replication. 20 However, if such episomes are to be used as a basis for the development of vectors for gene therapy applications, any chance of integration must be avoided. Nonintegration of episomal vectors is secured by a human “Scaffold or Matrix Attachment Region,” the S/MAR element.
The S/MARs are AT-rich sequences along the chromosomes of 300 bp to several thousand bp long, residing exclusively within the eukaryotic genomes, where they are distributed in a nonrandom fashion, not harboring consensus sequences or motifs of any kind. They are evolutionary conserved, present in gene families such as the human hemoglobin or the immunoglobulin genes, as well as in plants such as the Arabidopsis thaliana, and it appears that they participate in the packaging process of chromosomes in eukaryotic nuclei. 21
The S/MARs, as of their A-T-rich parts, have the propensity to undergo local strand dissociation, exhibiting long regions of extensive destabilization. This facilitates their specific attachment to the nuclear matrix so that the chromosomes are tethered onto the nuclear matrix in a dynamic way during the cell cycle, driving the organization of chromatin into structural domains within eukaryotic chromosomes and therefore, in the regulation of DNA functions, namely, gene expression and DNA replication. 22 The S/MAR’s natural role is that of boundary formation through chromatin opening and chromatin organization into functional domains.
The S/MAR element, which is incorporated into the episomal vectors for gene therapy of hemoglobinopathies, derives from the 5′ of the human cytocine β-interferon beta gene (IFNB1), and it was inserted into the prototype episomal vector pEPI-1, 23 by replacing the gene for the T4 antigen. The S/MAR element within vector pEPI-1 has been a significant advancement in the development of episomal vectors.
Maintenance of transgene expression as a function of the S/MAR was shown by early studies,23,24 as well as later studies, 25 and derives from the S/MAR’s propensity to acquire an open configuration. This state evokes the action of ubiquitous and specific transcription factors, which, in the presence of mitotic stability, can maintain long-term transgene expression.
Mitotic stability of vector pEPI-1 is based on pEPI-1 plasmid’s binding to the protein SAF-A and anchoring onto the nuclear matrix. 17 This allows for the pEPI-1 plasmid’s segregation during mitosis along with the chromosomes of the respective cells, thus acquiring mitotic stability, leading to cell genetic modification and long-term vector retention in the nucleus of the recipient cell.
Blocking vector integration is a major property of the S/MAR element as part of an episomal vector, and it is assured by placing the S/MAR within the transcription cassette “promoter CMV-eGFP-S/MAR-PolyA site” so that transcription of eGFP continues through the S/MAR element. Actions like deletion of the eGFP gene or termination of transcription before reaching the S/MAR by placing the poly-A site between the eGFP gene and the S/MAR lead to plasmid integration. Consequently, transcription through the S/MAR element is a prerequisite for nonintegration. 23 Blocking integration may not be a universal property for S/MARs, as at least one S/MAR, the one from the murine c-myc gene, could not block integration. 26
S/MAR-based episomal vectors have the capacity to maintain transgene expression, are resistant to integration, and are able to confer mitotic stability in vivo to modified cells that are under selection pressure. 27 However, over a decade ago, a high number of preclinical trials have been reported, using S/MAR carrying episomal vectors in vivo, with no mention of selection, on various diseases, notably in inherited retinal diseases, causing loss of vision. These vectors used in combination with nanoparticles and electric methods for their entry into the retina cells, have produced states of the disease cure and a hope for a safer and effective option for future treatment. 28 Furthermore, very recent publications presented the use of S/MAR vector technology in growing tissue. In particular, kidney organoids were generated derived from human iPSC, which bear the capacity of long-term ErythroPoietin secretion. Experiments are still needed to consolidate the data regarding the efficacy and safety of the system; nevertheless, this case of measurable efficacy effect of an S/MAR vector on a system of dividing cells is great progress. 29 Evidently, important advancements in the development and application of S/MAR-based, nonviral episomal vectors have been achieved and present a promise for gene therapy applications.
Disadvantages and advantages of the episomal vectors
Disadvantages
Considering that “episome delivery” is the system used to facilitate the entrance of the episome through the plasma cell membrane in a nonspecific manner, the S/MAR-based episomal vectors need a delivery system to help them enter the cells. This is a major limitation to nonviral, episomal expression vectors and still remains a challenge as a “clinically relevant means to achieve nuclear delivery of the DNA cargo,” is still not available. 30 The most widely used delivery systems for episomal vectors are based on physical methods (e.g., electroporation, sonoporation) or on chemical methods (nanoparticles, gold particles, etc.), with the nanoparticles presenting the best outcomes when used with RNA or small DNA molecules. There is intense investigation regarding this topic of nucleic acid delivery along with modifying parameters. 31
A major issue is the low level of plasmid establishment upon entrance into the nucleus and after reaching a place on the nuclear matrix. To obtain establishment, the plasmid must acquire the status of a replicon by a stochastic process, which allows it to replicate. This depends mainly on the specific nuclear compartment—nuclear matrix—the vector reaches after entry into the nucleus or is guided to by genomic sequences in the plasmid, such as an insulator element, like the chicken β-like globin gene—the cHS4. 32 As shown in subsequent sections, Section 5cii, chromatin-acting genetic elements may be the key to enhancement of vector establishment.
S/MAR-based episomal vectors may show toxicity due to the presence of bacterial genes of resistance to antibiotic used as selection markers during the culture of the prokaryotic host. This may be manifested as horizontal transfer of such genes and their possible entrance into strains of receptive plasmid of endogenous bacteria. Deletion of the bacterial genes of the episomal vectors that enter the preclinical trial state is the recommended and followed approach.
S/MAR-based episomal vectors are not expected to evoke strong immune response by the recipient subject as they do not carry any viral coding genes by design. 23 However, DNA sequences of viral origin within the episomal vectors, such as promoters, origins of replication, poly-A sites, and the dinucleotide Cytocine-phosphate-Guanine (CpG) areas, may cause an immune response, depending on the transgene, the backbone sequences, which can be replaced by a milder form or deleted.
Advantages
The nonviral, S/MAR-based, episomal vectors:
Are nonintegrative into the endogenous, genomic DNA, and this confers protection toward insertional mutagenesis/oncogenesis, an important drawback of the viral vectors. Are noninvasive for the genome, as they do not act on the genome in any way. Do not pose the problems encountered by the production of viral vectors, or similar to them, as of their plasmid nature, even in a large-scale production. Are fairly easy to handle in order to construct, manipulate, and use them, as they require only common molecular biology lab facilities. Are fairly easy to distribute to remote places, as they are made of DNA, which travels easily in dry ice. Finally, they are much less costly in general, an important parameter to take into consideration.
The S/MAR element from the 5′ of the β-interferon gene provides an excellent model for the inclusion of a strong S/MAR supporting nonintegration, transgene expression, and mitotic stability in an episomal vector.
GENETIC ELEMENTS OF THE β-GLOBIN LOCUS THAT ENHANCE THE FUNCTION OF S/MAR-BASED EPISOMAL VECTORS
Episomal vectors were originally based on viral genetic elements in order to induce cell modification with a stable level of transgene expression; for example, the system EBVoriP-EBNA-1. However, viral gene products can evoke immunological reactions and/or cause harmful interactions within the host cell. The idea slowly evolved that nonviral episomal vectors can be constructed of human, chromosomal, and genetic elements,32,33 if possible, entirely of such elements. Genetic elements, located within the β-globin genes locus, have been recognized as having a major impact on the native genes’ organization, transgene expression, and DNA replication. These are: (a) the β-globin cell-specific promoter and the spleen focus-forming virus (SFFV) ubiquitous and CD34+ cell-functional promoter; (b) the LCR, directing the regulation of expression of the β-globin genes locus; and (c) the replication Initiation Region (IR or β-globin Replicator), a bona fide mammalian origin of replication.
These entities will be presented here as pertinent to the design and development of S/MAR-based, efficient episomal vectors.
A cell-specific/cell-functional, strong promoter
Τhe promoter of the β-globin gene is erythroid tissue cell-specific. Eighteen different mutations (https://globin.bx.psu.edu/) have been recorded, from −28 to −101 bp, all related to β+/β+ type of β-thalassemia. These mutations are known to decrease the rate of transcription of the HBB gene due to decreased binding of transcriptional factors, leading to lower levels of mRNA transcripts. No β0 type of β-thalassemia has been reported so far, resulting from mutations located in the promoter of the β-globin gene.
The promoters of the β-like globin genes (ε-, γA-, γG-, δ-, and β-globin) prevail in the competition with their nearest ones for binding to the respective LCR site at the corresponding developmental stage of expression, and within this context they are all strong, high-potency promoters. This process is particularly important, considering that the expression of the β-globin genes is principally regulated at the stage of transcription, carried out by the combinatorial action of transcription factors. 34 Our current understanding of the combinatorial action of transcription factors is, in short, that they are binding to low-affinity sites of the promoter, allowing for high-frequency interactions. This occurs in cooperation with neighboring similar molecules, enabling the promoter-LCR enhancer system to activate the target gene in the right chromatin domain at the right time for gene expression. 34
The high-potency of the HBB gene promoter is supported and maintained by the presence of binding sites for a number of ubiquitous and specific transcription factors, including the ones for two major transcriptional regulators, namely: The DNA sequence CCACACCCT, for the binding of the Erythroid Krüppel-like factor (EKLF or KLF1), a hematopoietic-specific transcription factor that directly regulates the three-dimensional organization of the β-globin genes locus during terminal erythroid differentiation 35 and a consensus binding site for the DNA-binding protein CTCF (CCCTC-binding factor), a ubiquitously expressed nuclear protein, which plays a crucial role in the organization of the three-dimensional nuclear matrix and chromatin as well as in the transcriptional regulation in vertebrates.34,36
On the one hand, conditionally strong ubiquitous promoters for mammalian cells, tested as part of nonviral, episomal vectors, have shown that episomal transgene’s promoter is cell-dependent. 5 The viral (cytomegalovirus early) pCMV ubiquitous promoter is widely used with a wide spectrum of mammalian cells due to its ability to support highly efficient transcription of various genes. pCMV was included in the prototype episomal vector pEPI-1 to drive the transcription cassette “pCMV-eGFP-S/MAR_polyA”; however, it was silenced in progenitor CD34+ cells. 37
On the other hand, another ubiquitous promoter SFFV (from the spleen focus-forming virus), also a widely applied promoter of genes within mammalian cells, was found to be functional in CD34+ cells. Therefore, ubiquitous promoter SFFV replaced pCMV in driving eGFP transcription in pEPI-1 derived episomal vector, and it was effective as a CD34+ cell-functional promoter, with positive results. 25
We envisage a promoter-enhancer system of cooperating transcription factors functioning within an episomal vector that could define the potency and the tissue specificity of promoters, along with even more agents involved in the regulation of transcription of episomal genes. Interestingly, studies with the S/MAR-based episomal vector pEPI-GFP in stable transfections in mammalian cells revealed that the induction of chromatin changes, for example, DNA methylation, histone deacetylation, and more, either on the host or the vector, affects the nuclear localization of the episomal vector molecules as well as the transcriptional activity of the episomal transgenes, showing that their transcription is, at least partly, regulated at the chromatin level. 38
The β-globin-specific promoter and the ubiquitous SFFV promoter and functional in CD34+ cells provide a definite model for the incorporation of a strong cell-specific or cell-functional, transgene promoter within episomal vectors.
The LCR
The LCR is localized within the genetic locus of the “β-globin-like genes,” ∼50 kb 5′ of the HBB gene, extends over 20 kb and is comprised of five DNase I hypersensitive sites (HSs), HS1 to HS5 (Fig. 1A). Key function of the LCR is to act as a “transcriptional activator” in vivo for the genes within the β-globin genes locus, 39 and this operation of LCR is described as the most important level of regulation of the β-globin gene expression. However, when the LCR functions in vitro, for example, when it is transferred, along a β-globin gene into cells, transgenic animals, it acts as a “transcriptional enhancer” rather than an activator. This is because, in these conditions, the HBB gene is already performing a basal level of transcription, guided by the β-globin promoter and the enhancer located within the β-globin IVSII, and the addition of the LCR raises the transcription of the HBB gene to physiological levels.40,41 The function of the LCR, within integrated viral vectors, is not influenced by suppression phenomena from heterochromatin expansion, and hence, the term “super enhancer” has been attributed to this genetic element. 40

The genetic locus for the beta-like globin genes cluster.
The β-globin LCR carries additional, important properties, such as (a) regulation of developmental stage-specific expression of the β-globin genes, (b) chromatin remodeling to provide accessibility for each β-globin gene to the transcriptional machinery, 42 (c) insulation and barrier function to protect the β-globin genes from the repressive effects of surrounding heterochromatin, 43 and (d) long-range interactions via looping of the DNA to bring LCR sequences in close proximity to the proper β-globin gene at the proper developmental stage. This physical interaction is critical for the effective up-regulation of these genes.44,45 However, by forced rewiring, using the synthetic transcription factor ZF-Ldb1, the promoter-enhancer contacts in humanized transgenic mice bearing human “β-like globin genes” can change so that the newly formed LCR-HBG (γ-globin) constructs, when engrafted into host animals, may give rise to adult, erythroid cells with increased HBG, mimicking the Heretitary Persistence of Fetal Hemoglobin (HPFH) syndrome. 40
Transcriptional regulation of the β-globin genes via LCR was originally demonstrated by studies on β-thalassemias deriving from genomic deletions, encompassing this genetic area located a long distance (50 kb) 5′ of the β-globin gene cluster. These studies reported transcriptional inhibition of the β-globin genes locus associated with chromatin condensation as a result of the genomic deletions, such as the widely studied Dutch deletions or the Hispanic deletion.
3
The involvement of the LCR in the transcriptional regulation of the β-globin genes locus was initially revealed by studies on DNA deletions. In particular, the Hispanic deletion (Fig. 1B), a natural deletion upstream of the β-like globin genes, including the LCR sequence starting at HS2 site, extending up to HS5, and further 27 kb. This is, unexpectedly, associated with a complete lack of transcription of the γ-, δ-, and β-globin genes, appearing under the clinical phenotype of “γδβ0 thalassemia,” 46 while the γ-, δ-, and β-globin genes are present and intact within the genetic locus. 47 The Hispanic deletion is associated with changes in the pattern of DNA replication, deriving from the transition of the respective, permissive chromatin configuration into the nonpermissive state of inactive chromatin, surrounding the β-globin genes locus. Noticeably, research with transgenic mice has shown that experimentally introduced deletions of the LCR elements from HS2 to HS4 or from HS2 to HS5 recapitulate the clinical condition of Hispanic deletion thalassemia. 48 Studies on Hispanic deletion demonstrated the function of LCR as a transcriptional activator.
The “Hispanic deletion globin locus” is a natural model highlighting sequences located at a long distance from a gene as potential transcriptional regulatory elements.
The LCR consists of five DNase I HSs, which are indicative of open chromatin regions to promote accessibility to transcription factors. Combinations of core elements of DNase hypersensitive sites HS2, HS3, and HS4 were identified as supporting the highest levels of human β-globin expression per gene copy in viral gene transfer protocols. All three of them may act as an integral holocomplex unit to regulate the human HBB gene, while elements HS2 and HS3 combined can direct the strongest induction of transcription of β-globin genes, as revealed by systematic deletion mutagenesis in stable MEL transfectants. 49 In a more detailed analysis, the greater impact of element HS2 as a powerful transcriptional enhancer was verified in recent years. Furthermore, the study has shown that the element HS2 plays a role in the formation of the β-globin topological domain, 50 the important substructure within nuclear architecture, supporting the regulation of gene expression.
It follows that the LCR regulates transcription over a considerably long distance and, by general consensus, the LCR element mediates contact with their coordinate genes through the formation of intrachromosomal loops 51 (Fig. 1C). Such long-range gene regulation in vivo, involves spatial interactions of higher-order chromatin between transcriptional elements, with intervening chromatin looping out, and the formation of higher-order chromatin facilitated by the function of MAR elements, keeping chromatin onto the nuclear matrix. Noticeably, the β-globin LCR harbors MAR elements that exist in critical positions and function in vivo, named from 5′ to 3′ direction, MARHS4, MARHS2, MARε, and MARγΑ. 52 Core sequences of these MARs contribute to looping events by matrix attachment, engaging active genes of the human β-globin gene cluster and as such, they are involved in the regulation of β-globin-like genes’ transcription. 53
The clinical relevance of LCR function to the gene therapy of β-hemoglobinopathies has been confirmed by a plethora of such studies with viral gene transfer. Clinical trials have been reported on the ex vivo gene transfer of self-inactivating, lentiviral vectors carrying the physiological HBB gene or its antisickling version. These vectors were transferred into autologous CD34+ cells and resulted in tissue-specific and regulated expression of the HBB gene. In these vectors, the HBB gene was under its own promoter and core elements of the β-globin LCR were included. Lentiviral vectors used were the GLOBE), 54 carrying the elements HS2 and HS3 of the LCR; the BB305 vectors,55,56 carrying the LCR elements HS2, HS3, and HS4; and the lentiviral vector TNS9.3.55, 8 carrying the same elements as vectors BB305. The LCR, HS2, and HS3 elements included in these three vectors vary in size from one vector to the other, and so does the element HS4 between the vectors BB305 and TNS9.3.55. The efficacy and safety issues referring to ongoing trials with transfusion-dependent patients with β-thalassemia, using these three vectors, are discussed in a rather recent review. 57 The LCR included in the nonviral, episomal vectors so far is the micro-LCR,58,59 a 6.5 kb DNA encompassing all five HS elements of the β-globin LCR in their longer DNA sequence. Therefore, no conclusions can be drawn on their possible individual effect on a nonviral episomal vector’s performance, and only specific and focused studies will elucidate this issue. Suffice it to state that the micro-LCR has to be significantly shortened, if only because the total plasmid of these vectors has to be of much lower size in order to achieve a higher transfection efficiency.
HS sites and MAR elements within the LCR of the β-globin genes locus provide a model to show that the respective DNA sequences operating within higher-order chromatin domains facilitate transcription of the HBB gene.
The
β
-globin replication IR
The genetic element of replication IR, first described in the literature in 1993, 59 is localized at the 5′ of the HBB gene (Fig. 1D), and it defines the point of the β-globin initiation of physiological, bidirectional DNA synthesis within the human genome. Deletion of the IR sequence, as in the case of the Lepore syndrome, results in the loss of bidirectional DNA synthesis, replaced by replication in the upstream direction within the locus, and this constitutes the first finding, as a genetic proof, of the existence of specific element of IR in animal cells. 59 The IR was identified as a cis-acting DNA sequence, capable of inducing replication in parts of the genomic DNA that do not possess such capacity, 60 it is considered to be a bona fide mammalian Replicator, and it is named the β-globin Replicator.
The exact sequences that confer Replicator activity to the β-globin IR, have been specified in a detailed analysis of its sequences. The IR element includes a number of AT-rich parts occurring in disperse positions, as well as a DNA sequence of asymmetric purine:pyrimidine within a 300 bp essential core sequence (Fig. 1D). This sequence possesses the actual capacity for DNA replication, as tested by in vitro mutagenesis. 52 Efficient initiation of DNA replication by the IR requires the function of different combinations of an AT-rich DNA sequence with the asymmetrical purine:pyrimidine DNA sequence in the 300 bp essential track, with the first one defining the initiation site, whereas the second one enhances the frequency of replication initiation. 52
Studies on β-hemoglobinopathies, carrying the genomic “Lepore deletion,” are illuminating regarding the effect of the deletion of the IR on the function of the physiological β-globin genes.
The “Lepore deletion” (Fig. 1D) is a naturally occurring genetic condition within the β-globin gene locus that removes a DNA piece that includes the β-globin Replicator. This way the Lepore deletion cancels normal, bidirectional DNA synthesis, and then, a passive, unidirectional replication takes place, in the upstream direction only, from an external origin. These changes in the β-globin genomic DNA replication derive from changes in the chromatin organization caused by the DNA deletion; they lead to the silencing of the adult δ- and β-globin genes and are associated with δβ-hemoglobinopathy.
The Lepore deletion is considered to have derived from nonhomologous recombination, involving the δ-globin and β-globin genes. This process resulted in a deletion of 7.2 kb in the DNA of the respective patient, where the 5′ terminus of the deletion is in the δ-globin gene, but the 3′ terminus is in the β-globin gene, giving rise to the Lepore hybrid δβ-globin gene, causing the δβ-hemoglobinopathy or the “Lepore syndrome” (Fig. 1D). The anti-Lepore hybrid βδ-gene is also produced in the process and presents normal hematological phenotype. 61
The Lepore syndrome is a natural model to show the necessity of IR for normal, bidirectional DNA replication and the expression of the adult globin genes.
The IR element within nonviral, episomal vectors
The IRs, despite their identity as origins of DNA replication in mammalian cells, cannot, by themselves, support DNA replication when they are inserted into a plasmid or an episome and are passed into mammalian cells. In order to be effective in such conditions, they have to be linked to a viral replicon or chromatin-associating sequences of the genome. 62
The IR element, introduced into the study of episomal vectors rather recently, 63 derives from the genome region between 5226.995 and 5228.615 sites (HBB ENSG00000244734), or between the +50 and the −1,570 base pairs in connection to the β-globin transgene sequence.
Four studies, conducted thus far, implicating the IR as part of episomal, S/MAR-based vectors (Fig. 3) have demonstrated its dramatic, positive effects on various aspects of episome function.63–65
The IR enhances vector replication, as documented for the first time with the commercial plasmid pCEP4 (Fig. 2A.I), 63 which contains neither the S/MAR nor the IR element. Studies with plasmid pCEP4 were based on the formulation of stress-induced destabilization profiles, (SFFV), which estimate the energy needed for dissociation of every base pair in consecutive order within a circular DNA molecule. 66 The insertion of the S/MAR element into plasmid pCEP4 (Fig. 2A.II), surprisingly, was followed by the death of the respective cell culture under selection pressure, within a few days of transfection. This was thought to be attributed to a failure of plasmid replication, which deprived the cells of antibiotic resistance. The subsequent insertion of the IR into the episomal pCEP4, replacing the EBV OriP (Fig. 2A.III) was followed by the continuation of the cell culture, presumably because pCEP4 plasmid replication was resumed. Research based on SFFV profiles led to the conclusion that the IR generated the required helix destabilization, which released the torsional stress imposed by the S/MAR on the pCEP4 plasmid DNA sequence, thus rendering the plasmid accessible to the replication machinery. 63 Importantly, this study identified the IR as an origin of replication, ensuring sufficient replication to an episomal vector in the absence of any viral origin of replication.
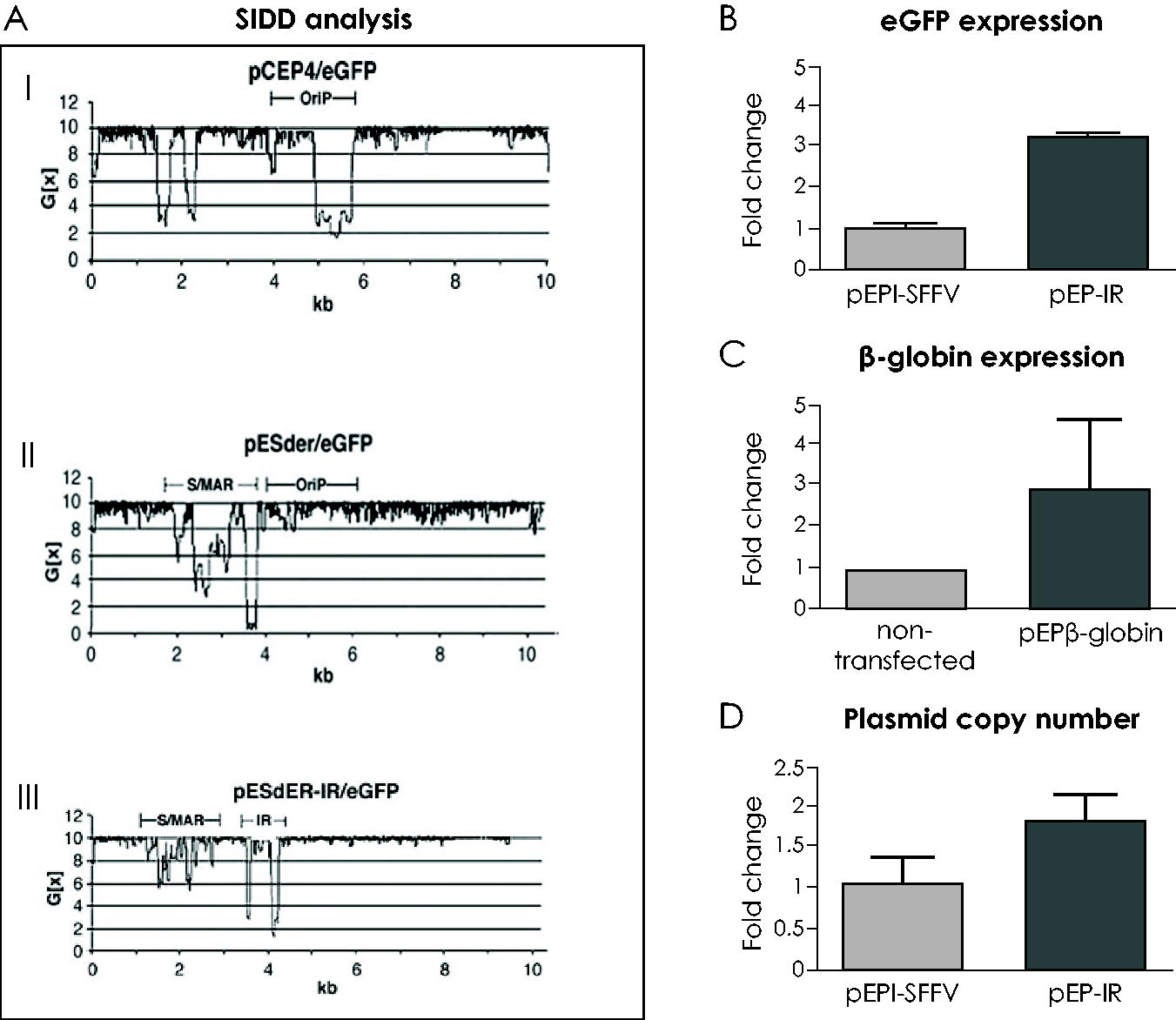
The effect of IR on replication and expression.
The IR also has a positive effect on transgene expression, but only on established plasmids. It supported a threefold increase in eGFP expression (Fig. 2B) per plasmid copy of vector pEP-IR (Fig. 3II) relative to the control vector pEPI/SFFV lacking the IR element (Fig. 3I). 25 These results were obtained from fluorescent, transfected cell colonies derived from a CFC assay of transfected CD34+/eGFP+ hematopoietic cells. Similarly, results obtained with vector pEPβ-globin (Fig. 3III), which includes the IR, documented a threefold increase in the HBB gene expression (Fig. 2C). This increase reaches the level of the adult HBB gene expression 67 ; however, its therapeutic value remains to be investigated and verified in preclinical and clinical trials.
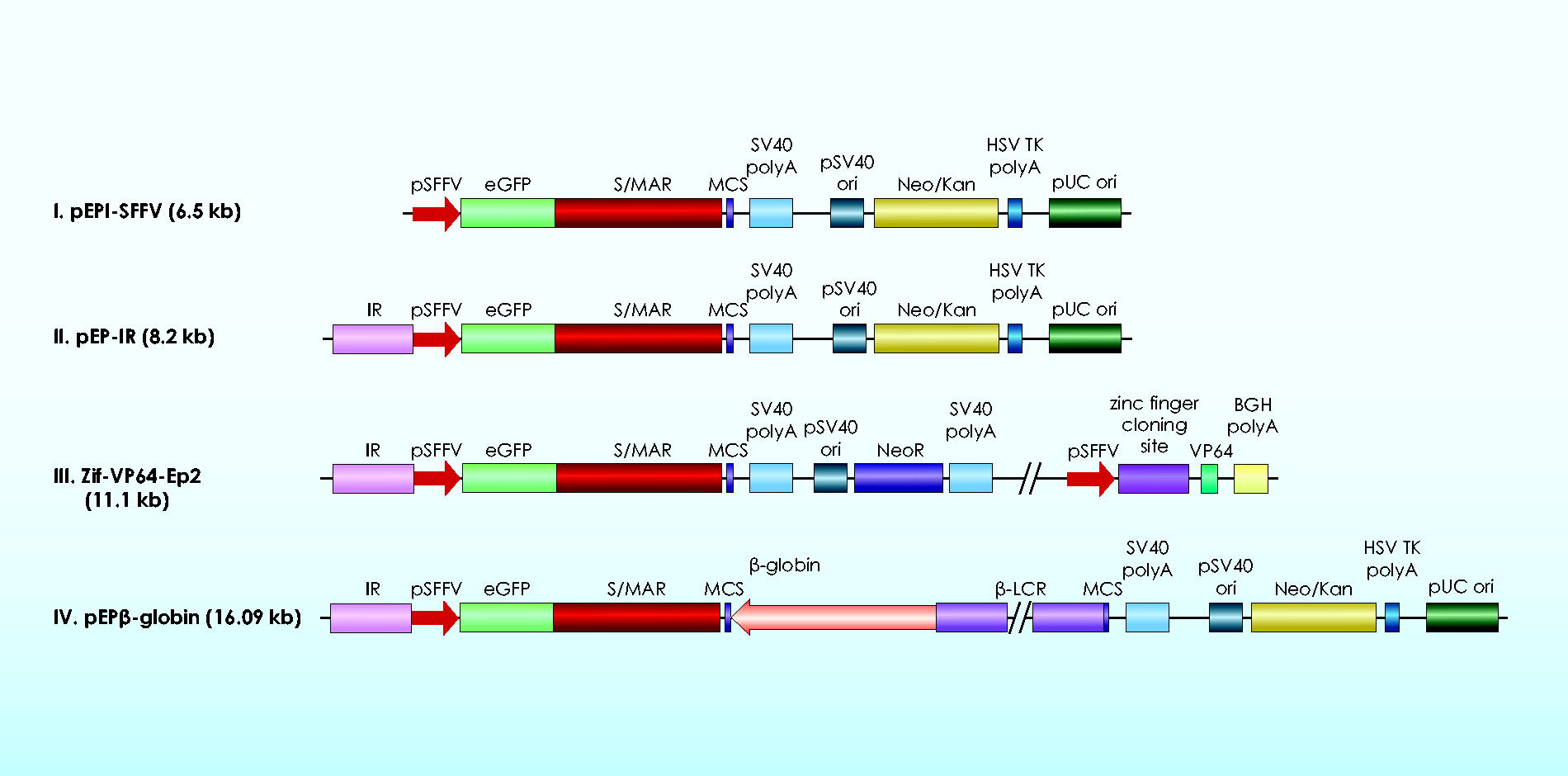
Constructs used for the study of the IR function.
The IR enhances vector establishment in transfected CD34+ cell derivatives, estimated as the mean percentage of fluorescent colonies over the total number of colonies derived from differentiated, descendant cells of the transfected CD34+/eGFP+ cells. Studies were carried out on the IR function within pEPI-1 derived vectors II, III, and IV (Fig. 3), which achieved a mean establishment of up to 93.76% fluorescent colonies (Table 1), in contrast to the control vector I, lacking the IR element, whose establishment was estimated to be 56.91% fluorescent colonies. 25 The IR is the only one out of the four genetic elements presented here that enhances vector establishment.
Results from Colony-Forming Cell Assay
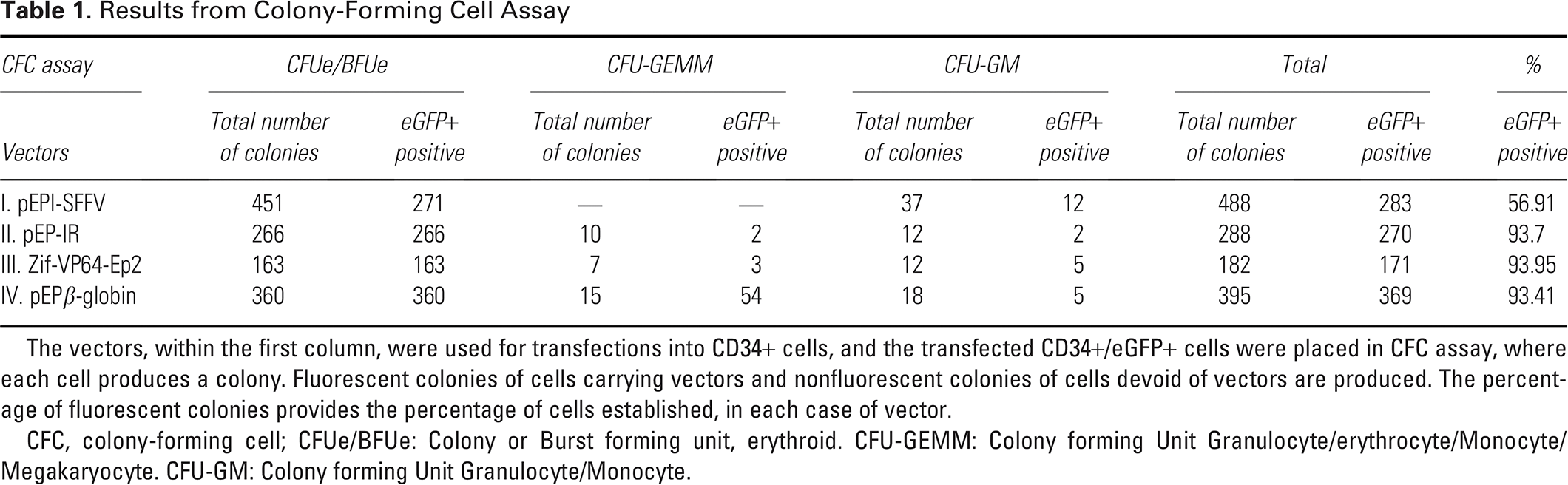
The vectors, within the first column, were used for transfections into CD34+ cells, and the transfected CD34+/eGFP+ cells were placed in CFC assay, where each cell produces a colony. Fluorescent colonies of cells carrying vectors and nonfluorescent colonies of cells devoid of vectors are produced. The percentage of fluorescent colonies provides the percentage of cells established, in each case of vector.
CFC, colony-forming cell; CFUe/BFUe: Colony or Burst forming unit, erythroid. CFU-GEMM: Colony forming Unit Granulocyte/erythrocyte/Monocyte/Megakaryocyte. CFU-GM: Colony forming Unit Granulocyte/Monocyte.
Previously reported enhancement of vector establishment by genetic elements within nonviral, episomal vectors, 32 involved the human, Ubiquitous Chromatin Opening Element A2UCOE and the β-globin chicken hypersensitive Site 4 (cHS4) element, in Chinese Hamster Ovary (CHO) cells. Despite the differences in the identity of the genetic elements studied and the cells used in transfections, a twofold increase in establishment against the control vector has been achieved in all reported cases. This confirms the idea that chromatin-acting genetic elements may be the key to the enhancement of vector establishment.
Finally, an increase in plasmid copy number per cell is supported by the presence of IR in episomal vectors, reaching mean values of 1.78, which is a 2.57- to 3.56-fold increase compared to vectors lacking the IR with mean values of 0.5–0.69 25 (Fig. 2D). The higher number of copy number of plasmids carrying IR can be attributed to the enhancement of plasmid replication and further improvement of episomal vector performance.
In short, the IR initiates replication in mammalian cells as the only origin of replication within the episomal vector (Fig. 2AIII), enhances plasmid establishment by nearly twofold (Table 1) and plasmid DNA replication by slightly over threefold increase in plasmid copy number per cell 25 (Fig. 2D), and raises transgene expression by a threefold increase in the transgene mRNA 64 (Fig. 2B and C).
The β-globin IR, emerging as a Replicator, provides an excellent model for the inclusion of a strong initiation of Replication within episomal vectors, supporting all critical aspects of their design, namely establishment, replication, and expression.
THE FUNCTIONAL COMPLEMENTARITY OF HUMAN GENETIC ELEMENTS WITHIN EPISOMAL VECTORS FOR β-HEMOGLOBINOPATHIES
The sine qua non condition for the design of an episomal vector to be used in preclinical trials is the successful completion of efficient transfection of the progenitor and stem cells CD34+ and the cell modification of the transfected CD34+ cells post stable transfection. These two requirements are connected to the two major drawbacks that are marking the function of S/MAR-based, nonviral, episomal vectors, namely, vector delivery and plasmid establishment. These two drawbacks render S/MAR-based episomal vectors, so far, much less efficient than viral vectors.
Nonviral, episomal vectors, as previously stated, need a delivery system to facilitate their way through the plasma membrane for their entrance into the cell, and this is a serious challenge for episomal vectors that directly affects transfection efficiency. The transfection efficiency of pEPI-derived, S/MAR-based episomal vectors, defined as the percentage of plasmid-bearing cells over total live cells, drops with increasing size of the episome. In recent studies with transfections of hematopoietic cells25,64,65 transfection efficiencies were rather low, around 36.8% and 24.62%, but they are, nevertheless, acceptable, considering the rather big size of the vectors (6.5–16.09 kb) (Fig. 3). Moreover, the electroporation (nucleofection) method for transfecting human hematopoietic progenitor cells results in very low rates of cell viability. Transfection efficiency has to be substantially improved for nonviral, episomal vectors to enter the route to clinical trials. To this end, improved delivery systems of vector DNA are essential, and the deletion of vector sequences, such as bacterial genes for antibiotic resistance, may be very helpful.
The very low level of cell modification due to the respective low level of establishment of these vectors onto the nuclear matrix of the recipient cell is another limitation. To obtain establishment, vectors must pass through a series of stages as presented in Section 4.
Considering the way the IR element mediates the increase of plasmid establishment, vectors carrying the chicken cHS4 insulator showed increased plasmid establishment in CHO cells, leading to the hypothesis that it could be due to interactions between the cHS4 insulator and nuclear matrix proteins. 32 In a later study, applying “4C technology of advanced, genome-wide analysis” of the contact sites of S/MAR-based replicons on the nuclear matrix in HeLa cells, it was documented that established vector molecules reside preferentially within actively transcribed regions (sites rich in RNA polymerase II) and also co-exist with DNA polymerases. 68 It is, therefore, reasonable to speculate that plasmids carrying an efficient IR have a greater chance of establishment in a contact site rich in RNA and DNA polymerases than plasmids devoid of the IR element.
Any increase in transfection efficiency (e.g., by a more efficient promoter or delivery system) leads to an increased number of episomal vectors landing on the nuclear matrix. On these vector-plasmids and at contact sites rich in DNA polymerase II, acts the IR element induces higher rate of plasmid establishment. Subsequently, the S/MAR element acts upon these established plasmids to induce a higher rate of cell modification, as the S/MAR acts only on established plasmids. The higher rate of cell modification thereof is the result of functional complementarity of the three genetic elements, the promoter, the IR, and the S/MAR, each of which acts upon an advanced background created by the genetic element acting at the preceding stage.
Furthermore, all these genetic elements, β-globin promoter—β-globin LCR, β-globin IR, and β-interferon S/MAR—have been shown to be capable of enhancing transgene expression in established plasmids. The addition of the LCR to a S/MAR-based episomal vector containing the HBB gene under its native promoter raises HBB gene transcription by twofold, 58 compared to untransfected cells, and by threefold to full physiological level upon addition of the IR element, 47 in stably transfected K562 cells. The functional complementarity among the LCR, the IR, and the S/MAR leads to higher levels of transgene expression.
Established, replicating, and expressing vector molecules may acquire mitotic stability by anchoring the genomic DNA onto the nuclear compartment in which they reside. The state of mitotic stability of the episomal vector, for example, pEPβ-globin, is provided principally by the 5′ interferon S/MAR element. The episomal vector cannot proceed to cell modification in the absence of an S/MAR element, 65 even though two MAR elements exist within the micro-LCR, 5′ of the HBB gene 58 bearing anchoring capacity, 65 but apparently cannot support vector mitotic stability in the absence of the interferon S/MAR element. The S/MAR element is thus emerging as a substantial, nuclear matrix anchoring element, capable of inducing cell modification as well as plasmid nonintegration to established episomal vectors.
Three genetic elements, β-globin LCR, β-globin IR, and β-interferon S/MAR, contain AT-rich regions along their sequence that may undergo local strand dissociation; they are all chromatin modulators that enhance interactions among them and between established plasmid DNA and nuclear matrix proteins, contributing to enhanced transgene expression and the mitotic stability of the vector.
Evidently, the aforementioned genetic elements act in functional complementarity with each other, performing specific interconnecting model functions, facilitating their individual contribution toward cell modification, and mediating their collective performance to achieve optimal HBB gene expression in episomal vectors.
DISCUSSION AND CONCLUSIONS
The development of nonviral episomal vectors for the gene therapy of inherited, monogenic disorders is within the context of gene addition strategy, as is the use of viral vectors for the same purpose.
In parallel, a number of other nonviral gene transfer systems have been developed for gene therapy applications. 5 Over 10 antibiotic-open-access vectors, which address the plasmid toxicity related to the presence of bacterial genes, are currently assessed in phase I/II clinical trials 69 and hopefully, will advance our understanding of how these safer plasmids fare within specific cellular environment. Nonviral gene transfer systems of great potential are DNA-based transposable elements, which integrate into the genomic DNA with the action of a transposase. A number of transposable elements are used for gene transfer into mammalian/human cells, particularly in primary human cells, presenting different functional efficiencies in preclinical and clinical trials. The relative transposition efficiency of these different transposon systems depends on various aspects, such as the cell type used for transfection, the transfection efficiency, the transposon/transposase ratio, the quality and quantity, and concentration of DNA used for transfection. Most widely studied of these systems is the Sleeping Beauty (SB) transposable element, which is also the most highly promising one, due to its high efficiency of transposition as well as to nearly random distribution of its insertion sites in the genome. SB, therefore, appears to satisfy to a great extent the efficacy as well as the safety issue in a unique way compared to other similar systems, such as the PiggyBac or the Tol2 systems, which present a less favorable safety profile as they are inserted mostly into genes and their regulatory sequences.70,71 The development of transposon-based systems shows that they advance ahead of the episomal vectors. However, none of the above nonviral systems of antibiotic-open-access vectors or transposable elements seem to have been applied to the HBB gene within the context of β-thalassemia. This may be due to the complexity of the regulation of expression of the β-globin genes, but also to the restriction in the size of the transgene load allowed in these systems. Furthermore, the parallel existence and operation of multiple gene transfer systems seem to be desirable, even necessary.
The β-hemoglobinopathies, characterized by the absence of DNA sequences or by the presence of point mutations in the β-globin genes locus both of which generate a “lack of gene function” pathology, form a complete and unique human disease model for the design of efficient nonviral, episomal vectors for gene transfer into cells of hematopoietic origin.
The lessons learned from studies of the hemoglobinopathies dictate that the design of nonviral, episomal vectors for gene therapy of human diseases must contain the human genetic elements involved in the in vivo regulation of transgene transcription. These genetic elements are as follows:
A CD34+ cell-specific or cell-functional, high-potency promoter for in vivo and in vitro studies respectively, guiding high rates of transgene transcription. A strong S/MAR element, capable of: (i) strong matrix attachment through binding to resident nuclear matrix proteins so as to facilitate long-term plasmid retention and stable cell culture, and (ii) blocking integration into the endogenous DNA, as in the case of the human S/MAR element 5′ IFNB1-gene by ensuring its constant transcription. A transcriptional enhancer, the LCR, acts to elevate the transgene transcription rate to physiological levels. A strong origin of replication, the β-globin replicator, for enhancement of all vector functions and in particular, plasmid replication and establishment. Importantly, the IR can ensure sufficient replication of an episomal vector composed of genomic sequences, even in the absence of any viral origin of replication.
The study on hemoglobinopathies allows for the prediction of modifications for wider clinical use, as it leads to the identification of crucial genetic elements for the regulation of gene expression and chromatin modulation. Such genetic elements may be applied in any combination considered suitable, depending mainly on the identity of the cells in which an episomal vector resides.
For example, the presence only of the genetic elements a and b within the minimal, nano-S/MAR DNA episomal vector is adequate in order to support cell modification and stable cell culture. This is a nonintegrating vector carrying only one eukaryotic transcription cassette, “pCMV-eGMP-S/MAR-poly-A site,” which was used to genetically modify patient-derived cells, with improved plasmid establishment, higher transgene expression, and episomal maintenance. 72
In the case of the hemoglobinopathies, the genetic elements that have been identified as supporting the episomal state of the vector within the transfected cell are considered to form associations with the nuclear matrix and with each other and to provide the vector with specific attributes, as necessary and adequate for vector efficiency. This allows the episomal vector to function at its best capacity. Furthermore, these elements provide auto-competence, facilitating the vector’s progression through the fundamental stages of its fate inside the transfected cell, namely, the establishment, replication and expression, mitotic stability, and equal plasmid segregation to daughter cells.
Collectively, these elements present a “model” demonstrating that genetic elements of high strength, encompassing a cell-specific or cell-functional promoter, a Replicator, and a matrix attachment region, acting in functional complementarity, are necessary and sufficient for the design of efficient episomal vectors for gene therapy applications. These vectors can be developed to offer a valid alternative to viral vector gene therapy.
AUTHORS’ CONTRIBUTIONS
A.A.: Conceptualization and writing—original draft. A.S.: Review and editing (equal). M.V.: Review and editing (equal).
Footnotes
ACKNOWLEDGMENTS
The support of the Laboratory of General Biology, Medical Faculty, University of Patras, to A.A. is gratefully acknowledged.
FUNDING INFORMATION
No funding was received for this article.
AUTHOR DISCLOSURE
The authors confirm that there are no relevant financial or nonfinancial competing interests to report.