
Abstract
Chronic kidney disease (CKD) is a major global health problem characterized by renal fibrosis, for which effective therapeutic options are still lacking. Mesenchymal stem cells (MSCs) have emerged as potential candidates for treating fibrosis due to their paracrine effects. This study first compared the antifibrotic capacities of umbilical cord-derived MSCs (UCMSCs) and dental pulp stem cells (DPSCs). The results showed that DPSCs exhibited superior effects in suppressing fibrosis markers and improving the fibrotic microenvironment. Thus, subsequent studies focused on DPSC and their hepatocyte growth factor (HGF)-modified counterpart (HGF-DPSC). Using an in vivo unilateral ureteral obstruction (UUO) mouse model and an in vitro Transforming Growth Factor-Beta 1(TGF-β1)-induced Human Renal Proximal Tubule Epithelial Cell (HK-2 cell) model, this study systematically evaluated the promising antifibrotic effects and mechanisms of DPSC. The results demonstrated that HGF-DPSC significantly improved the fibrotic microenvironment by regulating the Phosphoinositide 3-Kinase/Protein Kinase B/Glycogen Synthase Kinase 3 Beta (PI3K/AKT/GSK3β) signaling pathway and suppressing β-catenin activation. We confirmed direct protein–protein interaction between HGF and Iodothyronine Deiodinase 2 (DIO2) through co-immunoprecipitation (Co-IP), which suggested a novel molecular mechanism by which HGF-DPSC exerts its antifibrotic effects. These findings highlight the multitarget mechanism of HGF-DPSC in the treatment of renal fibrosis and provide new insights and possibilities for the treatment of CKD.
INTRODUCTION
Chronic kidney disease (CKD) is considered as one of the major global public health issues, with its prevalence and mortality rates continuing to rise. 1,2 Renal fibrosis, a hallmark pathological feature of CKD, 3 –5 is commonly observed as the end-stage manifestation of various kidney diseases, including diabetic nephropathy, 6 acute kidney injury, 7 and chronic glomerulonephritis. 4 It is characterized by tubular epithelial cell (EC) injury, 8 inflammatory cell infiltration, 9 fibroblast activation, 10 excessive extracellular matrix (ECM) deposition, 11 and irreversible destruction of renal architecture. 12 As fibrosis progresses, kidney function gradually deteriorates, eventually leading to renal failure and associated complications, which severely impact patients’ quality of life and survival. 13
Given the current lack of effective therapeutic approaches to reverse fibrosis, there is an urgent need to explore novel strategies to mitigate fibrosis progression and promote renal tissue repair. 14 Current treatments for renal fibrosis primarily focus on symptomatic management, such as controlling hypertension, 15 regulating metabolic disorders, 16 and reducing inflammation. 9 However, these methods cannot effectively reverse established fibrosis or repair damaged tissues.
Mesenchymal stem cells (MSCs) have gained increasing attention in regenerative medicine and cell therapy due to their multipotent differentiation ability, immunomodulatory properties, 17 and low immunogenicity. 18 Studies indicate that the therapeutic potential of MSCs largely depends on their paracrine activities. 19 By secreting various bioactive factors, MSCs can modulate the pathological microenvironment, inhibit excessive ECM deposition, suppress inflammatory responses, and promote tissue repair. 20 However, significant differences exist among MSCs from different sources in terms of paracrine factor profiles, antifibrotic capacity, and tissue repair potential. 21
Umbilical cord-derived MSCs (UCMSCs) have been extensively studied due to their abundant availability, low immunogenicity properties, and stable proliferative capacity. 22 UCMSCs can reduce ECM deposition and regulate the fibrotic microenvironment by secreting anti-inflammatory factors. 23,24 In contrast, compared with UCMSCs, dental pulp stem cells (DPSCs) showed stronger anti-inflammatory and antifibrotic properties. 25 DPSCs can be collected noninvasively and possess higher proliferative capacity and stronger antioxidant properties. At P6, intracellular Reactive Oxygen Species (ROS) production was significantly less in DPSC culture in comparison to UCMSC. The β-gal expression also showed significantly low expression in DPSC. 26 Studies have shown that DPSCs secrete paracrine factors such as hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), and basic fibroblast growth factor 2 (FGF-2), which exhibit stronger regulatory effects in antifibrosis and tissue repair. 27 These factors can more effectively inhibit excessive ECM deposition, reduce fibroblast activation, and repair damaged cells.
Paracrine factors, particularly those modulating the fibrotic microenvironment, play a pivotal role in fibrosis progression. 28 Among them, HGF has been identified as a multifunctional paracrine factor with essential roles in antifibrosis and tissue repair. 29 HGF can inhibit fibroblast activation, reduce ECM deposition, and promote proliferation and migration of renal tubular ECs, thereby significantly improving renal function and tissue structure. 30,31 However, endogenous HGF expression is markedly reduced in the fibrotic microenvironment, limiting its therapeutic potential. 32,33 To overcome this limitation and enhance the antifibrotic efficacy of MSCs, this study employed an adenoviral vector (Ad.HGF) to genetically modify MSCs, thereby increasing HGF secretion and optimizing their paracrine signaling. 34
In this study, an in vitro HK-2 cell fibrosis model was utilized to compare the antifibrotic effects of UCMSCs and DPSCs. 35 Additionally, an in vivo unilateral ureteral obstruction (UUO) mouse model was established, and the enhanced effects of HGF modification on DPSCs-mediated antifibrotic activity were evaluated. 36,37 Furthermore, transcriptome sequencing and validation experiments were performed to elucidate the potential mechanism of HGF-DPSC in regulating fibrosis, providing insights into the application of MSCs gene modification in the treatment of renal fibrosis.
MATERIALS AND METHODS
HK-2 cell culture and establishment of an in vitro fibrosis model
Human renal proximal tubular ECs (HK-2), purchased from ATCC, were cultured in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a 5% CO2 incubator. The culture medium was replaced every 2 days, and cells were passaged when reaching 80–90% confluency using 0.25% Trypsin-Ethylenediaminetetraacetic Acid (EDTA).
To establish an in vitro fibrosis model, HK-2 cells were treated with TGF-β1 (10 ng/mL). Specifically, HK-2 cells were seeded at a density of 5 × 105 cells/well in six-well plates and cultured until reaching 70% confluency. After serum starvation for 12 h to synchronize the cell cycle, TGF-β1–containing serum-free medium was added, and cells were incubated for 24 h to induce fibrosis. Subsequently, conditioned media (CMs) derived from UCMSC, DPSC, or HGF-modified DPSC were added, and cells were incubated for an additional 24 or 36 h. Cells were harvested for downstream analyses, including quantitative real-time PCR (qRT-PCR), Western blotting, and immunofluorescence (IF) assays.
Expansion of DPSC and UCMSC and preparation of HGF-modified MSCs
Clinical-grade DPSCs and clinical-grade UCMSCs, provided by Beijing SH Biotechnology and China Medical Management Consulting, were cultured in serum-free medium. Cells were initially seeded at a density of 8,000 cells/cm2 and maintained at 37°C in a 5% CO2 incubator. Once cells reached 80–90% confluency, they were passaged using 0.25% Trypsin-EDTA.
HGF modification was achieved using the adenoviral vector Ad.HGF. When DPSCs and UCMSCs were expanded to the fourth passage, they were seeded into six-well plates at 70% confluency. Ad.HGF was added at a multiplicity of infection of 100 in serum-free medium without antibiotics. After 12 h, the medium was replaced with complete medium, and cells were incubated for 48 h. Modified cells and their CMs were collected for subsequent experiments.
Animal model establishment and intervention
C57BL/6J male mice (6–8 weeks old, 20–25 g) were purchased from GemPharmatech Experimental Animal Co., Ltd. The mice were housed under controlled environmental conditions, including a 12 h light/dark cycle, a humidity level of 50–60%, and a stable temperature of 21–23°C. They had ad libitum access to food and sterilized water. After a 1 week acclimation period, the mice were randomly assigned to the following groups: control group (sham operation, n = 12), UUO model group (n = 12), DPSC treatment group (n = 12), and HGF-DPSC treatment group (n = 12). To establish the renal fibrosis model, UUO surgery was performed. The mice were anesthetized with 1% pentobarbital sodium (30 mg/kg) via intraperitoneal injection. A dorsal incision was made to expose the left ureter, which was proximally ligated near the renal pelvis using double sutures. In the control group, the ureter was isolated but not ligated. Immediately after surgery (day 0) and on day 7, DPSC or HGF-DPSC (1 × 106 cells/200 μL PBS) was injected into the tail vein. On day 14, kidney tissues and blood samples were collected. In each group of 12 mice, 3 were used for histopathological evaluation, 3 for transcriptome sequencing, and the remaining 6 for molecular experimental analysis. The mice were euthanized by intraperitoneal injection of an overdose of pentobarbital sodium.
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) under the ethical approval number IACUC-DWZX-2023-557.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. The concentration and purity of RNA were assessed using a NanoDrop One UV spectrophotometer (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized using the PrimeScript RT Reagent Kit (YiSheng Biotechnology). qRT-PCR was performed using a SYBR Green qPCR Kit (YiSheng Biotechnology) on a Bio-Rad real-time PCR system (Bio-Rad). Gene expression was normalized to Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) and expressed as relative messenger RNA (mRNA) levels. The detailed primer sequences are shown in Table 1.
Reverse Transcription Quantitative Polymerase Chain Reaction Primers
Each reaction was performed in a total volume of 20 μL, containing 10 μL SYBR Green mix, 2 μL primer mix (0.2 μM), 1 μL cDNA template, and 7 μL nuclease-free water. The thermal cycling conditions were as follows: 95°C for 3 min, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. Melting curve analysis was performed to confirm the specificity of amplification. Relative gene expression levels were calculated using the 2−ΔΔ Ct method, with GAPDH as the reference gene.
Western blot analysis
Protein extraction was performed using RIPA lysis buffer (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors. Protein concentration was determined using a BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples were denatured by mixing with 5 × Sodium Dodecyl Sulfate (SDS) sample loading buffer and heating at 95°C for 10 min. Equal amounts of protein from each group were loaded and separated on 10% SDS-polyacrylamide gels prepared using the Epizyme Gel Preparation Kit. SDS-Polyacrylamide Gel Electrophoresis (PAGE) was performed at 80 V through the stacking gel and 120 V through the resolving gel. Proteins were transferred onto 0.45 μm Polyvinylidene Difluoride (PVDF) membranes using a wet transfer system (Bio-Rad) at 250 mA for 2 h.
Membranes were blocked with 5% nonfat milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 h at room temperature, followed by overnight incubation at 4°C with the primary antibodies diluted as follows: α-SMA (1:1000, CST, 19245S); COL1A1 (1:1000, Abcam, ab34710); TGF-β1 (1:1000, Abcam, ab179695); E-cadherin (1:1000, Thermo Fisher Scientific, 13-1700); Vimentin (1:1000, Abcam, ab92547); HGF (1:1000, BOSTER, MA00089); DIO2 (1:1000, Abcam, ab77779); PI3K (1:1000, CST, 4292S); p-PI3K (1:1000, ABclonal, AP0427); AKT (1:1000, CST, 4691S); p-AKT (1:1000, CST, 2965S); GSK3β (1:1000, ABclonal, A2081); p-GSK3β (1:1000, CST, 5558S); β-catenin (1:1000, ABclonal, A20221); GAPDH (internal control, 1:5000, Abcam, ab8245); β-tubulin (internal control, 1:5000, Sungene Biotech, KM9003).
After washing with TBST (4 × 10 min), membranes were incubated with HRP-conjugated secondary antibodies (Goat anti-Rabbit, 1:5000, Sungene Biotech, LK2001 or Goat anti-mouse, 1:5000, Sungene Biotech, LK2003) for 1 h at room temperature. Chemiluminescent detection was performed using an ECL kit (Vazyme), and bands were visualized using the Tanon imaging system.
Small interfering RNA-mediated knockdown of HGF in DPSC
HGF expression in DPSCs was suppressed using specific small interfering RNA (siRNA) targeting the HGF gene (siHGF1, F: GCUAAUAGAUGUACUAGGA(dT), R: UCCUAGUACAUCUAUUAGC(dT); siHGF2, F: GGAGUUCCAUGAUACCACA(dT), R: UGUGGUAUCAUGGAACUCC(dT); siHGF3, F: CUCAGUGUUCAGAAGGUAA(dT), R: UUACCUUCUGAACACUGAG(dT); QingKe Biotechnology). Negative control siRNA was used as a control. DPSCs were seeded into six-well plates at a density of 6 × 105 cells/well and cultured to 50–60% confluency. SiRNA transfection was performed using Lipofectamine RNAiMAX Transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. After 6 h of transfection, the medium was replaced with complete medium containing 10 % FBS. Cells were harvested after 48 h for downstream analyses, including qRT-PCR and Western blotting, to verify the knockdown efficiency.
Immunohistochemistry of CKD biopsies
Kidney biopsy samples from patients with CKD were fixed in 4% paraformaldehyde for 24 h, followed by routine dehydration, clearing, and paraffin embedding. Sections (4 μm) were prepared, deparaffinized with xylene, and rehydrated through graded ethanol. Antigen retrieval was performed by heating sections in citrate buffer (pH 6.0) using a pressure cooker for 15 min. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide for 10 min, followed by blocking with 10% goat serum for 30 min at room temperature.
Sections were incubated overnight at 4°C with primary antibodies against DIO2 (1:200) and Collagen I (1:200). After washing with Phosphate-Buffered Saline (PBS), sections were incubated with HRP-conjugated secondary antibodies (1:500) for 30 min at room temperature. Staining was visualized using a DAB substrate, and counterstaining was performed with hematoxylin. After dehydration and mounting, slides were scanned using a digital scanner, and staining results were analyzed.
Immunohistochemistry of mouse kidney tissues
Mouse kidney tissues were harvested immediately after surgery and fixed in 4% paraformaldehyde for 24 h. The tissues were then processed for routine paraffin embedding, and 4 μm sections were prepared. The sections were baked at 60°C for 2 h, deparaffinized with xylene, and rehydrated through a series of graded ethanol solutions (100%, 95%, 85%, and 70%). Antigen retrieval was performed in citrate buffer (pH 6.0) using a pressure cooker for 15 min. After cooling to room temperature, the sections were rinsed with PBS three times (5 min each).
Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 min at room temperature. Nonspecific binding was minimized by incubating the sections in 10% goat serum for 30 min at room temperature. Primary antibodies (e.g., α-SMA, Collagen I, E-cadherin, vimentin) were applied to the sections at a dilution of 1:200, and the sections were incubated overnight at 4°C in a humidified chamber. After washing three times with PBS, the sections were incubated with HRP-conjugated secondary antibodies (1:500) for 30 min at room temperature.
Color development was achieved using DAB substrate solution under microscopic observation, with reaction times typically between 3 and 5 min to avoid overstaining. The sections were counterstained with hematoxylin, rinsed in running water for 15 min, dehydrated through graded ethanol, and cleared with xylene. The slides were mounted with neutral resin and observed under an Olympus microscope.
Immunofluorescence
HK-2 cells were seeded at a density of 1 × 105 cells/mL on sterile glass coverslips in 24-well plates and cultured until reaching 50–60% confluency. Cells were fixed with 4% paraformaldehyde for 15 min and washed three times with PBS. Fixed cells were permeabilized with 0.1% Triton X-100 for 10 min, followed by three washes with PBS. Nonspecific binding sites were blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature.
After blocking, the cells were incubated overnight at 4°C with primary antibodies (e.g., α-SMA and Collagen I) diluted at 1:200 in antibody dilution buffer. The next day, the cells were washed three times with PBS, and secondary antibodies conjugated with fluorescent dyes (1:500) were added. The cells were incubated at room temperature in the dark for 1 h. Nuclei were counterstained with 4′,6-Diamidino-2-Phenylindole (1:5000) for 10 min, followed by three PBS washes. Coverslips were mounted onto glass slides using an antifade mounting medium and allowed to dry at room temperature in the dark.
IF images were captured using a Zeiss confocal laser scanning microscope. The fluorescence intensity and localization of target proteins were analyzed, and differences between experimental and control groups were compared.
Co-immunoprecipitation
HEK293T cells were co-transfected with Hemagglutinin (HA)-tagged HGF and FLAG Epitope Tag (DYKDDDDK) (FLAG)-tagged DIO2 expression plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. After 48 h, cells were lysed in IP lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM sodium chloride [NaCl], 1% Nonidet P-40 (NP-40), 0.5% sodium deoxycholate) supplemented with protease inhibitor cocktail MedChemExpress (a commercial compound supplier) (MCE). Cell lysates were incubated with anti-HA or anti-FLAG antibody-conjugated agarose beads (Sigma-Aldrich) overnight at 4°C with gentle rotation. The beads were washed four times with wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% NP-40), and the immunoprecipitated proteins were eluted by boiling in SDS sample buffer. The samples were analyzed by Western blotting using anti-HA and anti-FLAG antibodies to detect HGF and DIO2, respectively.
Transmission electron microscopy
Mouse kidney tissues were collected, cut into small pieces (∼1 mm3), and immediately fixed in 2.5% glutaraldehyde (prepared in PBS buffer, pH 7.4) at 4°C for 6 h. The tissues were then washed three times with PBS buffer and postfixed in 1% osmium tetroxide (OsO4) at room temperature for 2 h. After postfixation, the samples were dehydrated through a graded ethanol series (50%, 75%, 95%, and 100%), with each step lasting 15 min, followed by further dehydration in pure acetone. The dehydrated samples were infiltrated with and embedded in epoxy resin (SPI-Pon™ 812, Structure Probe, Inc.) under vacuum conditions and polymerized at 60°C for 48 h. Ultrathin sections (70–90 nm thick) were prepared using an ultramicrotome (UC7, Leica Microsystems) and mounted on copper grids (300 mesh, Electron Microscopy Sciences). The sections were stained with 2% uranyl acetate (Electron Microscopy Sciences) for 10 min, rinsed with distilled water, and counterstained with 0.3% lead citrate (Electron Microscopy Sciences) for 5 min. Excess stain was removed by rinsing the grids with distilled water, and the sections were air-dried at room temperature. Ultrastructural observations were performed using a transmission electron microscopy (TEM; HITACHI HT7800, Hitachi Ltd.), focusing on podocyte foot processes, basement membrane thickness, and mitochondrial morphology.
Molecular docking and binding energy calculation
Protein structures were obtained from the UniProt database, 38 with full-length AlphaFold 39 -predicted models selected for DIO2_MOUSE (UniProt ID: Q9Z1Y9) as the ligand and HGF_HUMAN (UniProt ID: P14210) as the receptor. Molecular docking was performed using the HDOCKlite v1.1 server, 40 a highly efficient tool integrating physics-based and bioinformatics-based approaches for predicting biomolecular interactions. Global docking was carried out to sample all possible binding modes, and binding poses were ranked using scoring functions.
The model with the lowest binding energy (Model_1) showed a docking score of −288.19 and a confidence score of 0.9407, indicating a highly reliable binding mode. Binding free energy was further calculated using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method on the HawkDock server, 41 yielding a binding free energy of −55.68 kcal/mol. The binding mode was visualized using PyMOL, 42 and detailed interaction analyses, including key amino acid residues and binding sites, were performed to interpret the molecular interactions.
Bioinformatics analysis
For RNA-seq sequencing data, Bowtie2 43 software was used to align the clean reads to the SILVA database 44 to remove ribosomal RNA (rRNA) after removing low-quality sequences. HISAT2 software 45 was then used to align the rRNA-depleted reads to the reference genome. FeatureCount 46 was used to perform gene-level quantification for each sample, and the gene expression matrix for all samples was generated. Gene differential expression analysis was performed using edgeR. 47 KEGG enrichment analysis was conducted using the KOBAS (v3.0) software. 48 Cellanneal 49 was used for deconvolution of bulk RNA-seq data, using the public dataset GSE119531 50 as a reference, which is single-cell sequencing data from the UUO model. Gene regulatory network analysis was performed using https://genemania.org/. Gene Set Enrichment Analysis (GSEA) analysis was conducted using http://genepattern.org.
Statistical analysis
All experimental data are presented as mean ± standard deviation. Statistical analysis for comparisons among three or more groups was performed using one-way analysis of variance, followed by Bonferroni’s post hoc test for pairwise comparisons. A p value <0.05 was considered statistically significant. All analyses were conducted using GraphPad Prism version 9.0.
RESULTS
DPSC CM demonstrates superior antifibrotic efficacy compared to UCMSC CM in an in vitro HK-2 cells fibrosis model
Due to the variability in the composition and function of secretion factors of stem cells from different sources, screening stem cell sources with higher antifibrotic potential and exploring strategies to enhance their antifibrotic effects represent critical areas of investigation. Our previous work has proven that adenovirus expressing human HGF (Ad-HGF) transduction increased the expression of HGF in MSCs, 51,52 thereby optimizing their paracrine function and enhancing their antifibrotic effects. 53
In the present study, human proximal tubular ECs (HK-2) were employed to establish a fibrosis model induced by TGF-β1 stimulation, and then the antifibrotic potential of two different sources of MSCs, UCMSCs and DPSCs, and the enhancing effects of HGF modification was evaluated. Following model establishment, CMs from UCMSCs, DPSCs, and their HGF-modified counterparts were applied to assess changes in the expression of fibrosis-related genes and proteins. This approach was designed to identify stem cell sources with superior antifibrotic potential and to explore the mechanisms underlying the enhanced effects of HGF modification (Fig. 1A).
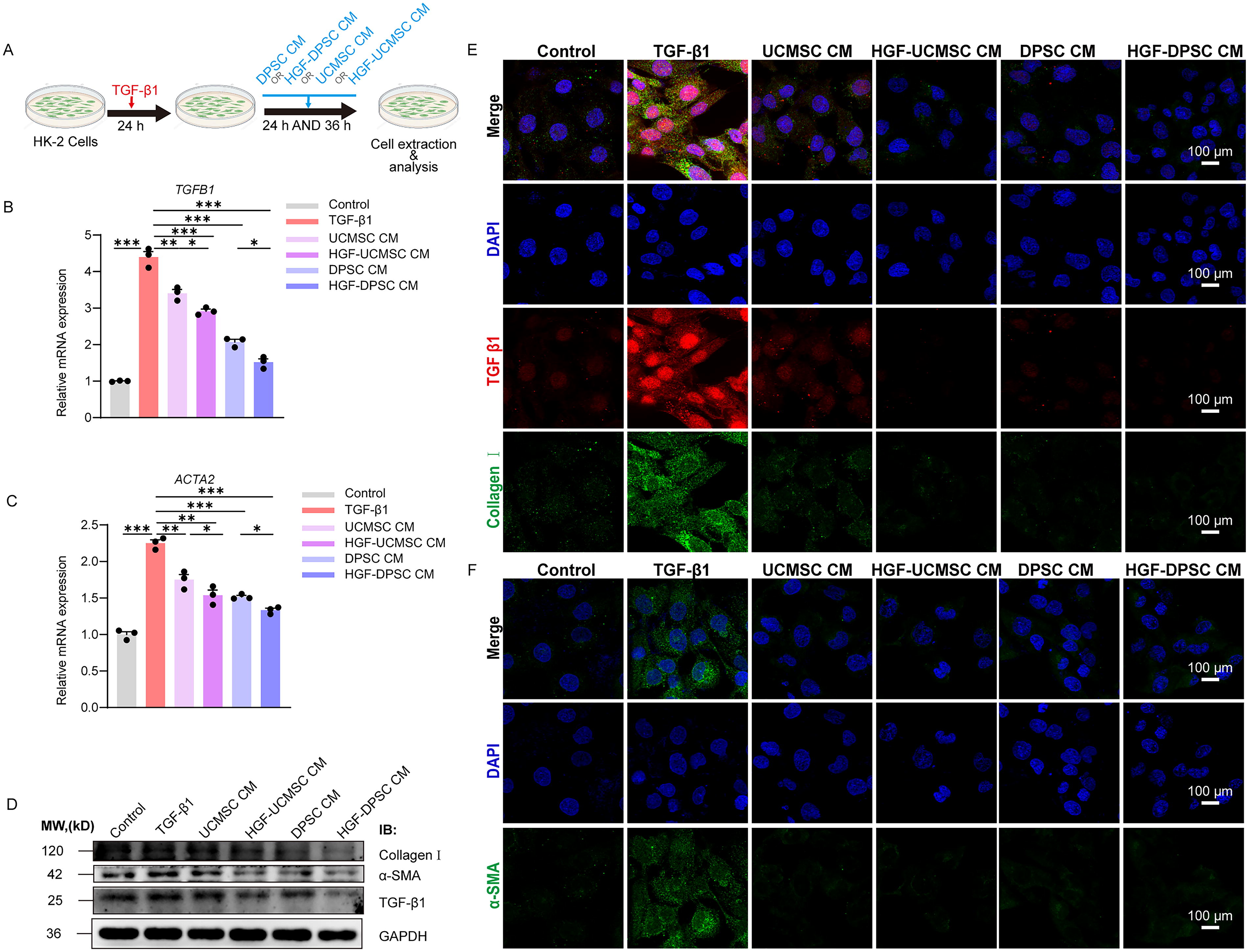
Antifibrotic effects of UCMSC, DPSC, and their HGF-modified conditioned media in a TGF-β1–induced HK-2 cell fibrosis model.
The results of qRT-PCR analysis revealed that the TGF-β1–induced model group exhibited a significant upregulation in the mRNA expression of fibrosis markers TGFB1 (Fig. 1B) and ACTA2 (encoding α-SMA) (Fig. 1C) compared to the control group (p < 0.001), confirming the successful establishment of the fibrosis model. Treatment with UCMSC CM and DPSC CM resulted in a marked reduction in the expression of TGFB1 and ACTA2 (p < 0.001), indicating that both stem cell sources possess antifibrotic properties. Notably, CM derived from HGF-modified stem cells demonstrated enhanced inhibitory effects, with HGF-DPSC CM showing the most pronounced suppression of TGFB1 and ACTA2 expression (p < 0.001). These findings suggest that paracrine factors secreted by stem cells significantly inhibit TGF-β1–induced fibrosis, and HGF modification further amplifies this antifibrotic capacity by augmenting paracrine activity.
Western blot analysis (Fig. 1D) corroborated the gene expression results. TGF-β1 significantly increased the expression of fibrosis-related proteins, including Collagen I and α-SMA, whereas both UCMSC CM and DPSC CM markedly reduced their expression. IF staining (Fig. 1E and F) provided visual confirmation of the expression and localization of fibrosis markers. In the TGF-β1–induced fibrosis model, the expression of Collagen I (Fig. 1E, green) and α-SMA (Fig. 1F, green) was significantly elevated and widely distributed. Following treatment with UCMSC CM and DPSC CM, the fluorescence signals for Collagen I and α-SMA were markedly attenuated. Furthermore, HGF-modified CM exhibited stronger inhibitory effects on the expression of TGF-β1, with the HGF-DPSC CM group displaying the weakest signal intensity, closely resembling that of the control group. Though the differentially expressed Collagen I and α-SMA between treatment groups in IF images are subtle, quantitative analysis of protein expression through Western blotting revealed distinct differences in treatment efficacy between DPSC CM and HGF-DPSC CM groups. The combined analysis of Western blot and IF data demonstrates that DPSC CM exhibit superior antifibrotic effects compared to UCMSC CM, and HGF modification further enhances this therapeutic efficacy.
HGF-DPSCs exhibit significant antifibrotic effects in the UUO mouse model
Renal fibrosis is a key pathological feature of CKD, characterized by renal dysfunction, interstitial fibrosis, and tubular atrophy. To evaluate the therapeutic potential of DPSC and their HGF-modified counterpart (HGF-DPSC) in the treatment of renal fibrosis, a UUO mouse model was established, which is a classical in vivo model for studying renal fibrosis. In our previous in vitro experiments, DPSC CM demonstrated superior antifibrotic efficacy compared to UCMSC CM, with HGF-modified DPSC CM (HGF-DPSC CM) showing the most significant inhibitory effects on fibrosis marker expression. Thus, in in vivo study, we focused on validating the antifibrotic effects of DPSC and HGF-DPSC.
In the experimental design, UUO surgery was performed on day 0, and DPSC or HGF-DPSC (1 × 106 cells/200 μL PBS/mouse) were immediately administered via tail vein injection after recovery from anesthesia. A second injection was given on day 7. On day 14, the mice were sacrificed, and kidney and blood samples were collected to assess therapeutic effects (Fig. 2A). The control group consisted of sham-operated mice without UUO.

Therapeutic effects of DPSC and HGF-DPSC on renal fibrosis in the UUO mouse model.
The results demonstrated that the kidneys in the UUO group showed significant swelling, which was reduced after treatment with DPSC or HGF-DPSC, with the HGF-DPSC group showing the most significant improvement (Fig. 2B). The mice in the UUO group exhibited significant renal dysfunction, as indicated by markedly elevated levels of blood urea nitrogen (BUN) (Fig. 2C, p < 0.001) and serum creatinine (CRE) (Fig. 2D, p < 0.001), confirming severe kidney damage induced by UUO. Treatment with DPSC or HGF-DPSC significantly reduced both BUN and CRE levels compared to the UUO group (p < 0.01). Statistical analysis revealed no significant difference between DPSC and HGF-DPSC groups in BUN reduction (p > 0.05). However, HGF-DPSC treatment resulted in significantly greater improvement in CRE levels compared to DPSC treatment alone (p < 0.05), indicating enhanced therapeutic efficacy of HGF-modified DPSCs in improving certain aspects of renal function. qRT-PCR analysis revealed that the expression of fibrosis markers Fn1 (encoding fibronectin 1) (Fig. 2E) and Acta2 (encoding α-SMA) (Fig. 2F) was significantly upregulated in the UUO group (p < 0.001). Both DPSC and HGF-DPSC treatments significantly downregulated these genes compared to the UUO group (p < 0.01). Direct comparison between treatment groups revealed that HGF-DPSC exhibited a significantly stronger inhibitory effect than DPSC alone for both Fn1 (p < 0.05) and Acta2 (p < 0.01) expression, highlighting the enhanced antifibrotic capacity conferred by HGF modification. Western blot analysis further confirmed these results (Fig. 2G). In the UUO group, fibrosis-related proteins such as TGF-β1, α-SMA, and vimentin were significantly upregulated, while E-cadherin was markedly downregulated, reflecting characteristics of renal fibrosis and epithelial–mesenchymal transition (EMT). Both DPSC and HGF-DPSC treatments effectively reversed these aberrant protein expressions, with HGF-DPSC producing the most significant improvements.
Histological analysis revealed severe structural damage in the kidneys of the UUO group (Fig. 2H). Hematoxylin and Eosin (H&E) staining showed significant tubular atrophy and extensive interstitial inflammation in the UUO group, while Masson’s trichrome staining indicated a marked increase in fibrotic area. Quantitative analysis revealed significantly higher renal injury scores (Fig. 2I) and Masson-positive areas (Fig. 2J) in the UUO group compared to the control group (p < 0.001). Treatment with DPSC or HGF-DPSC significantly reduced these scores and fibrotic areas compared to the UUO group (p < 0.001). HGF-DPSC treatment demonstrated significantly greater efficacy than DPSC treatment in reducing both renal injury scores (p < 0.05) and fibrotic areas (p < 0.01), providing histological evidence for the superior therapeutic effects of HGF-modified DPSCs.
Furthermore, TEM provided additional insights into the role of DPSC and HGF-DPSC in repairing renal ultrastructure (Fig. 2K and L). In the UUO group, extensive disappearance of podocyte processes, significant mitochondrial swelling, and cristae disruption were observed. Following treatment with DPSC or HGF-DPSC, these abnormalities were markedly improved, with HGF-DPSC showing the most significant restorative effects.
Transcriptomic analysis reveals the potential molecular mechanisms of HGF-DPSC treatment in UUO
To explore the molecular mechanisms underlying the therapeutic effects of HGF-DPSCs in UUO-induced renal fibrosis, transcriptomic profiling of kidney tissues from the control, UUO model, DPSC treatment, and HGF-DPSC treatment groups was conducted. Bulk RNA sequencing (RNA-Seq) was employed to identify differentially expressed genes (DEGs), revealing key molecular and cellular changes associated with fibrosis and therapeutic intervention.
Principal component analysis (PCA) was conducted to evaluate the overall transcriptomic differences among the experimental groups (Fig. 3G). The UUO model group showed a clear separation from the control group in PCA space, reflecting significant transcriptomic alterations associated with fibrosis.
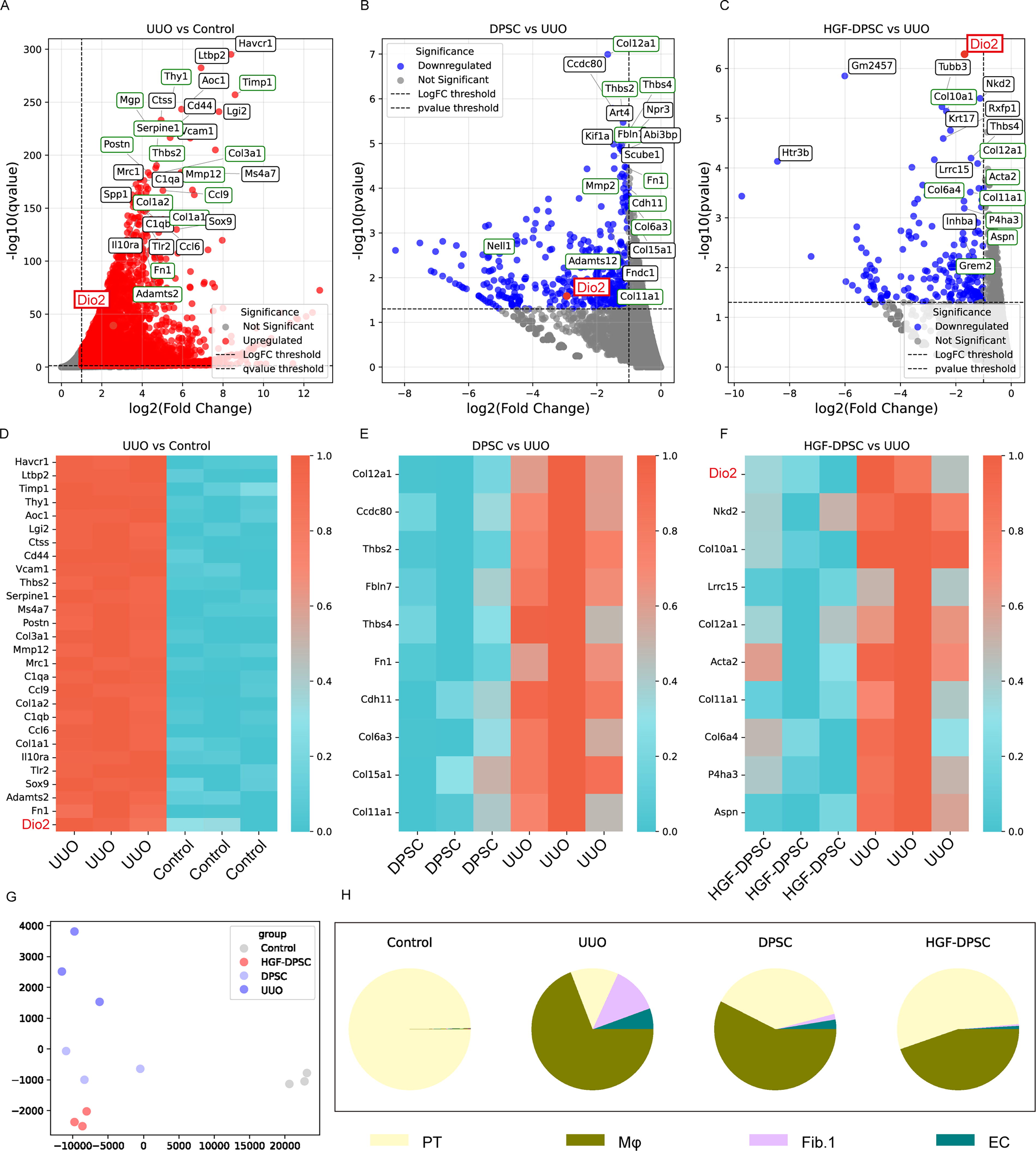
Transcriptomic analysis reveals gene expression changes in the UUO model and the regulatory effects of DPSC and HGF-DPSC treatments.
DEG analysis revealed significant transcriptomic alterations among the groups. In the UUO group, 6,952 DEGs were identified compared to the control group, including 4,216 upregulated and 2,736 downregulated genes (Fig. 3A). Among the upregulated genes, fibrosis-related markers such as Fn1, Col1a1, and Col3a1 were prominently elevated, as highlighted by green boxes in Fig. 3A, indicating excessive ECM deposition. Dio2 (highlighted in red) was also significantly upregulated in the UUO group compared to the control group. Pro-inflammatory genes, including Timp1 and Ccl6, were also upregulated, reflecting an inflammatory microenvironment characteristic of UUO-induced renal fibrosis. In the DPSC treatment group, 837 DEGs were identified compared to the UUO group (456 upregulated and 381 downregulated; Fig. 3B). Notable fibrosis- and inflammation-associated genes, including Col12a1, Fn1, Col6a3, Mmp2, and Ccdc80, were downregulated, suggesting that DPSC treatment attenuates fibrosis-related transcriptomic changes. The analysis also revealed that Dio2 (highlighted in red in Fig. 3B) was among the downregulated genes in the DPSC treatment group. Similarly, in the HGF-DPSC treatment group, 439 DEGs were identified compared to the UUO group (240 upregulated and 199 downregulated; Fig. 3C). Among these, fibrosis-related genes such as Col1a1, Col11a1, and Tgfb1 were significantly downregulated. Dio2 (highlighted in red in Fig. 3C) emerged as the most significantly downregulated gene following HGF-DPSC treatment. This gene has not been associated with renal fibrosis in previous studies, and its significant downregulation following HGF-DPSC treatment adds a new dimension to the understanding of fibrosis-related molecular mechanisms.
Heatmap analysis provided further insights into gene expression patterns across the experimental groups (Fig. 3D–F). Figure 3D shows that fibrosis-related genes, including Fn1, Col1a1, and Adamts2, as well as Dio2 (highlighted in red), were highly expressed in the UUO group compared to the control group. Figure 3E illustrates that DPSC treatment downregulated key fibrosis-associated genes such as Col12a1, Thbs4, and Ccdc80. Figure 3F demonstrates that HGF-DPSC treatment further downregulated fibrosis-related genes, including Col10a1, Col2a1, Col1a1, Acta2, and particularly Dio2 (highlighted in red), indicating significant modulation of the transcriptomic landscape in response to HGF-DPSC treatment and further supporting the potential role of Dio2 as a novel therapeutic target in renal fibrosis.
To investigate cellular composition changes, cell deconvolution analysis was performed using the publicly available single-cell dataset GSE119531, derived from UUO models, to infer the proportions of major kidney cell types (Fig. 3H). In the control group, proximal tubular cells (PT) dominated, with negligible contributions from macrophages (Mφ), fibroblasts (Fib.1), and ECs. In the UUO model group, PT was markedly reduced, while Mφ and Fib.1 increased, reflecting a fibrotic and inflammatory microenvironment. DPSC treatment partially restored PT proportions, accompanied by a reduction in Mφ and Fib.1 populations and a slight increase in EC. Notably, HGF-DPSC treatment led to a more pronounced reduction in Mφ and Fib.1 compared to DPSC treatment, with PT proportions showing a further recovery and EC levels slightly increasing. These findings suggest that while both treatments contribute to modulating the fibrotic microenvironment, HGF-DPSC treatment appears to have a stronger effect in restoring a healthy cellular composition, particularly by more effectively reducing fibrosis- and inflammation-associated cell populations.
HGF as a central paracrine mediator of the antifibrotic effects of DPSC
To elucidate the key role of HGF in the antifibrotic function of DPSC, siRNA was employed to specifically knock down HGF expression, and its impact on antifibrotic efficacy was evaluated. Three siRNAs (siHGF-1, siHGF-2, and siHGF-3) targeting HGF were designed (Fig. 4A). qRT-PCR (Fig. 4B) and Western blot (Fig. 4C) analysis revealed that siHGF-1 and siHGF-2 significantly reduced the mRNA and protein levels of HGF, whereas the knockdown efficiency of siHGF-3 was insufficient (< 70%). Therefore, siHGF-3 was excluded from further experiments, and siHGF-1 and siHGF-2 were utilized to ensure result reliability.
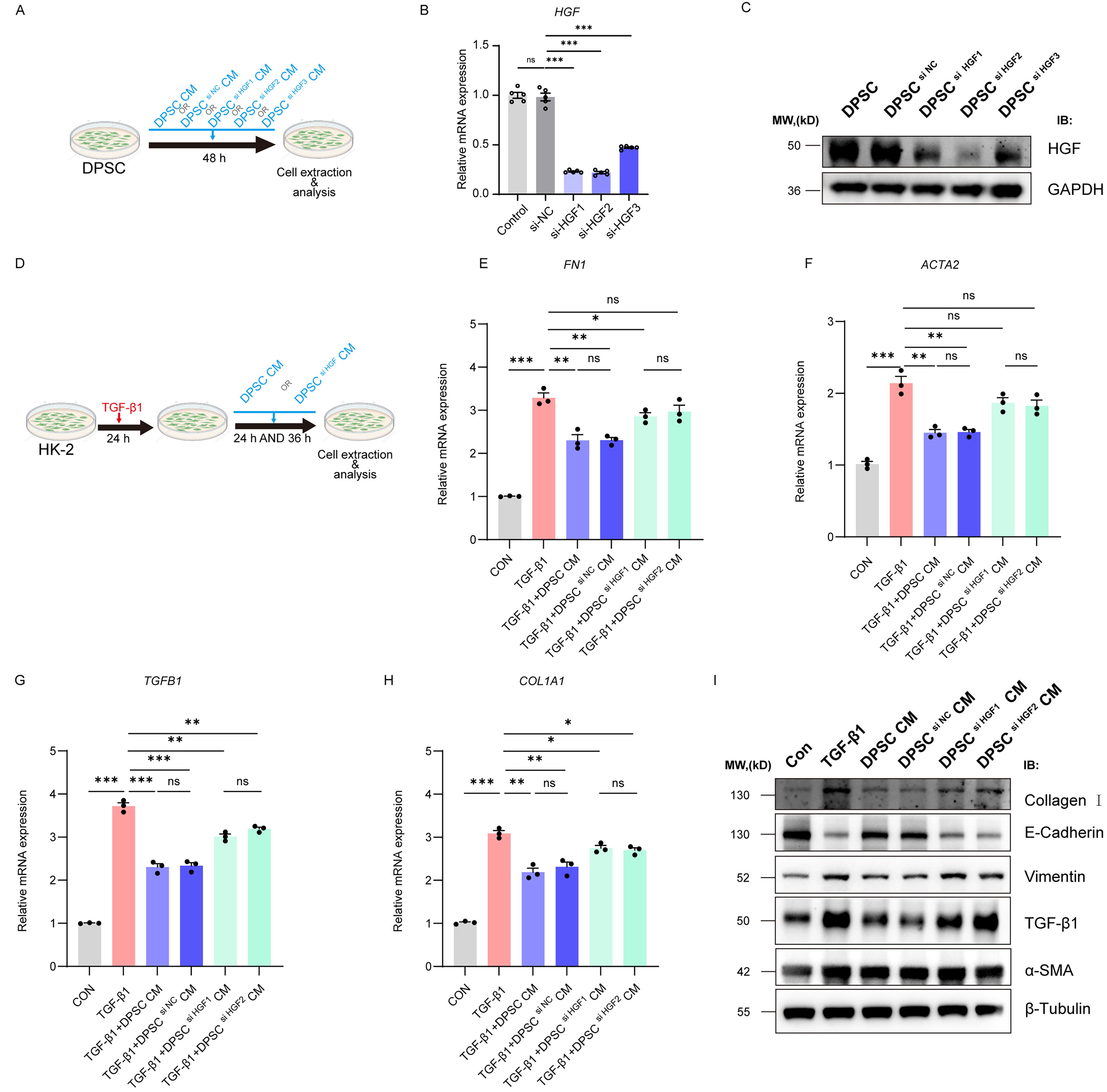
Critical role of HGF in the antifibrotic effects of DPSC-conditioned media in a TGF-β1–induced HK-2 cell fibrosis model.
In the TGF-β1–induced HK-2 cell fibrosis model, DPSC CM and DPSC si HGF CM were applied to assess their respective antifibrotic effects. HK-2 cells were pretreated with TGF-β1 (10 ng/mL, 24 h) to induce EMT, followed by exposure to DPSC CM or DPSC si HGF CM for an additional 36 h (Fig. 4D). qRT-PCR results demonstrated that TGF-β1 significantly upregulated the mRNA expression of fibrosis-associated genes FN1, ACTA2, TGFB1, and COL1A1 (Fig. 4E–H). DPSC CM significantly reduced the expression levels of these markers, whereas DPSC si HGF CM failed to achieve comparable suppression, showing no statistical difference from the TGF-β1 group (ns). These results underscore the indispensable role of HGF in the antifibrotic effects of DPSC CM.
Western blot analysis confirmed these findings at the protein level (Fig. 4I). TGF-β1 treatment elevated the expression of mesenchymal markers α-SMA, vimentin, TGF-β1, and Collagen I while downregulating E-cadherin. DPSC CM reversed these abnormal changes, whereas DPSC si HGF CM exhibited diminished effects, showing no significant improvement over the TGF-β1 group.
HGF-DPSC exerts antifibrotic effects through regulation of the PI3K/AKT/GSK3β-DIO2 signaling axis
To further explore the molecular mechanisms underlying the antifibrotic effects of HGF-modified DPSC (HGF-DPSC), we combined transcriptomic analysis with protein-level validation, focusing specifically on the PI3K/AKT/GSK3β signaling pathway. KEGG pathway enrichment analysis revealed a significant upregulation of the PI3K/AKT pathway in the UUO model group compared to the control group (Fig. 5A), suggesting abnormal activation of this pathway during the progression of fibrosis. Further GSEA analysis (Fig. 5B) confirmed the significant enrichment of the PI3K/AKT pathway in the UUO group, emphasizing its potential involvement in kidney fibrosis. In the DPSC treatment group, compared to the UUO model group, there was a significant downregulation of the PI3K/AKT pathway (Fig. 5C), indicating that DPSC treatment partially mitigates the aberrant activation of this pathway. GSEA analysis (Fig. 5D) further demonstrated a marked downregulation of the PI3K/AKT pathway in the DPSC treatment group, providing additional evidence that DPSC treatment contributes to modulating the fibrotic microenvironment. In the HGF-DPSC treatment group, the PI3K/AKT pathway was also significantly downregulated compared to the UUO model group (Fig. 5E), suggesting that HGF modification enhances the regulatory effect of DPSC on the fibrotic microenvironment. This therapeutic effect of HGF-DPSC on the PI3K/AKT pathway was further confirmed by GSEA analysis (Fig. 5F).
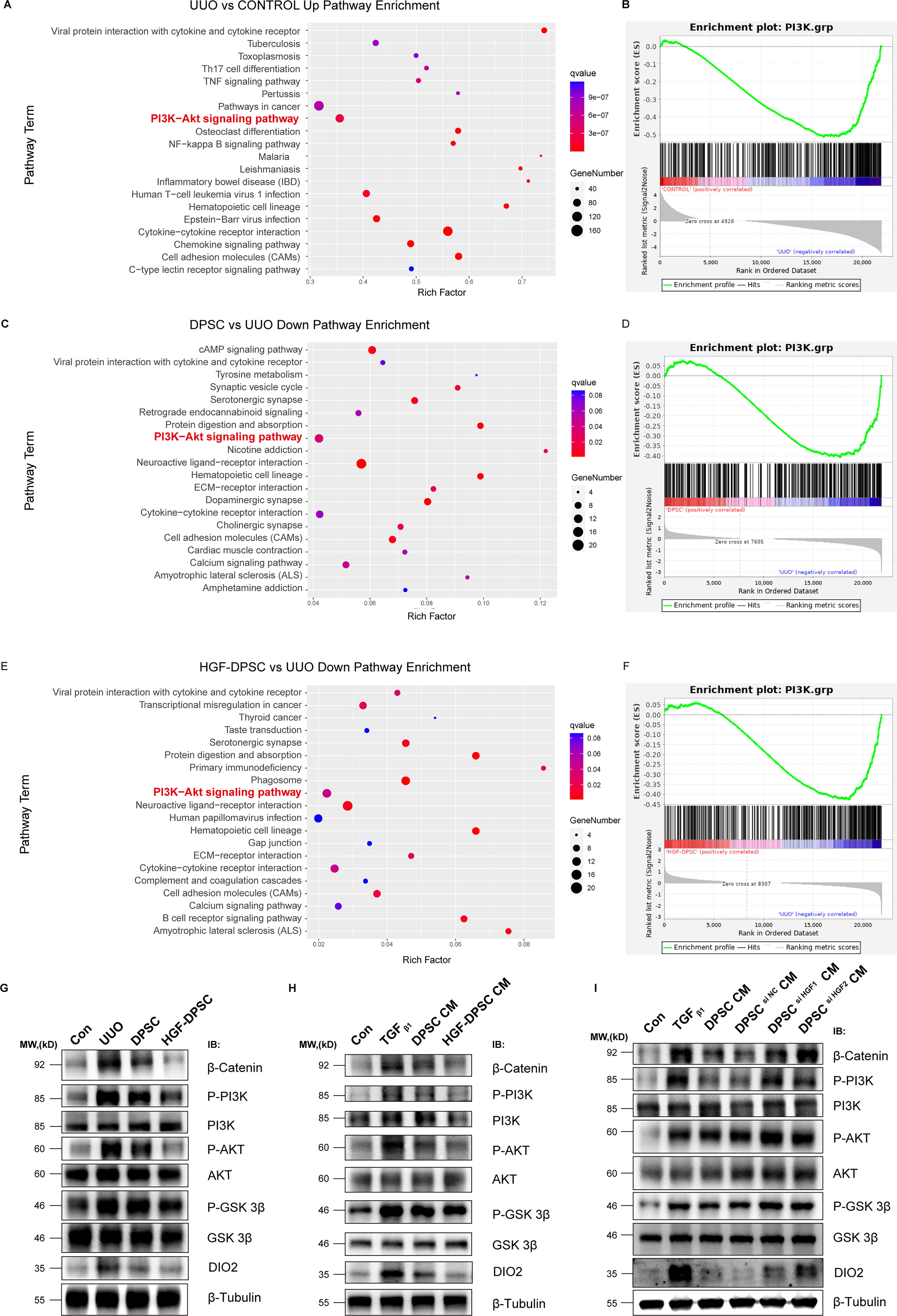
HGF-DPSC exerts antifibrotic effects through regulation of the PI3K/AKT/GSK3β-DIO2 signaling axis.
The PI3K/AKT signaling pathway is a key pathway in the fibrosis process, regulating cell proliferation, survival, GSK3β activity, and the aberrant expression of fibrosis-related genes, including β-catenin and DIO2. To validate these findings, we conducted Western blot analysis to assess the expression and phosphorylation levels of key proteins in the PI3K/AKT/GSK3β pathway and to examine DIO2 expression. Western blot results from both the UUO mouse model and the TGF-β1-induced HK-2 cell fibrosis model are shown in Fig. 5G and H. In both models, the phosphorylation levels of PI3K and AKT were significantly elevated, indicating abnormal activation of the PI3K/AKT pathway within the fibrotic microenvironment. Meanwhile, the phosphorylation levels of GSK3β also showed an increasing trend, suggesting enhanced activity, which leads to nuclear translocation and activation of β-catenin. Notably, DIO2 protein expression was markedly upregulated in both fibrotic models, displaying expression patterns that paralleled the changes observed in the PI3K/AKT/GSK3β pathway components. In the DPSC or DPSC CM treatment groups, the phosphorylation levels of PI3K and AKT were partially reduced, while GSK3β activity was significantly inhibited, leading to a reduction in β-catenin activation. Concurrently, DIO2 expression was partially downregulated. In the HGF-DPSC or HGF-DPSC CM treatment groups, the phosphorylation levels of PI3K and AKT were further decreased, and phosphorylation of GSK3β was more strongly suppressed, resulting in β-catenin activation levels that were nearly identical to those of the control group. Importantly, DIO2 expression was almost completely normalized in the HGF-DPSC treatment groups, suggesting a coordinated regulation between the PI3K/AKT/GSK3β pathway and DIO2 expression.
To further confirm the critical role of HGF in DPSC-mediated antifibrotic effects, we examined the effect of CM from DPSC with HGF knockdown (DPSC si HGF CM) on the PI3K/AKT/GSK3β pathway and DIO2 expression (Fig. 5I). The results showed that, compared to normal DPSC CM, DPSC si HGF CM was less effective in inhibiting the phosphorylation of PI3K and AKT. The suppression of GSK3β activity was also significantly reduced, leading to a substantial increase in β-catenin activation. Critically, knockdown of HGF in DPSC significantly impaired its ability to suppress DIO2 expression, with DIO2 levels remaining elevated in the DPSC si HGF CM group. These findings ultimately confirm that HGF is a critical factor mediating the antifibrotic effects of DPSC, primarily through the regulation of PI3K/AKT/GSK3β-DIO2 signaling axis.
DIO2 expression is significantly upregulated in human renal fibrosis samples
Previously, transcriptomic analysis of the UUO mouse model unexpectedly revealed that iodothyronine deiodinase (Dio2) was significantly upregulated in the UUO group and markedly downregulated following DPSC and HGF-DPSC treatment. These findings suggest that DIO2 may play a critical role in the development and treatment of renal fibrosis. Currently, there are no specific reports documenting the upregulation of DIO2 in renal fibrosis or kidney disease. DIO2, an enzyme that activates thyroid hormones by converting T4 into T3, is widely expressed in various tissues. To investigate whether this finding is consistent in human renal fibrosis, the Nephroseq database (https://www.nephroseq.org) was utilized to analyze the expression of DIO2 in renal tissues of patients with CKD (Fig. 6A). The database analysis revealed that DIO2 expression was significantly elevated in CKD renal tissues compared to normal renal tissues (p < 0.05, fold change = 2.733). Building on these findings, further validation of DIO2 expression changes during fibrosis progression was carried out using immunohistochemical (IHC) analysis.
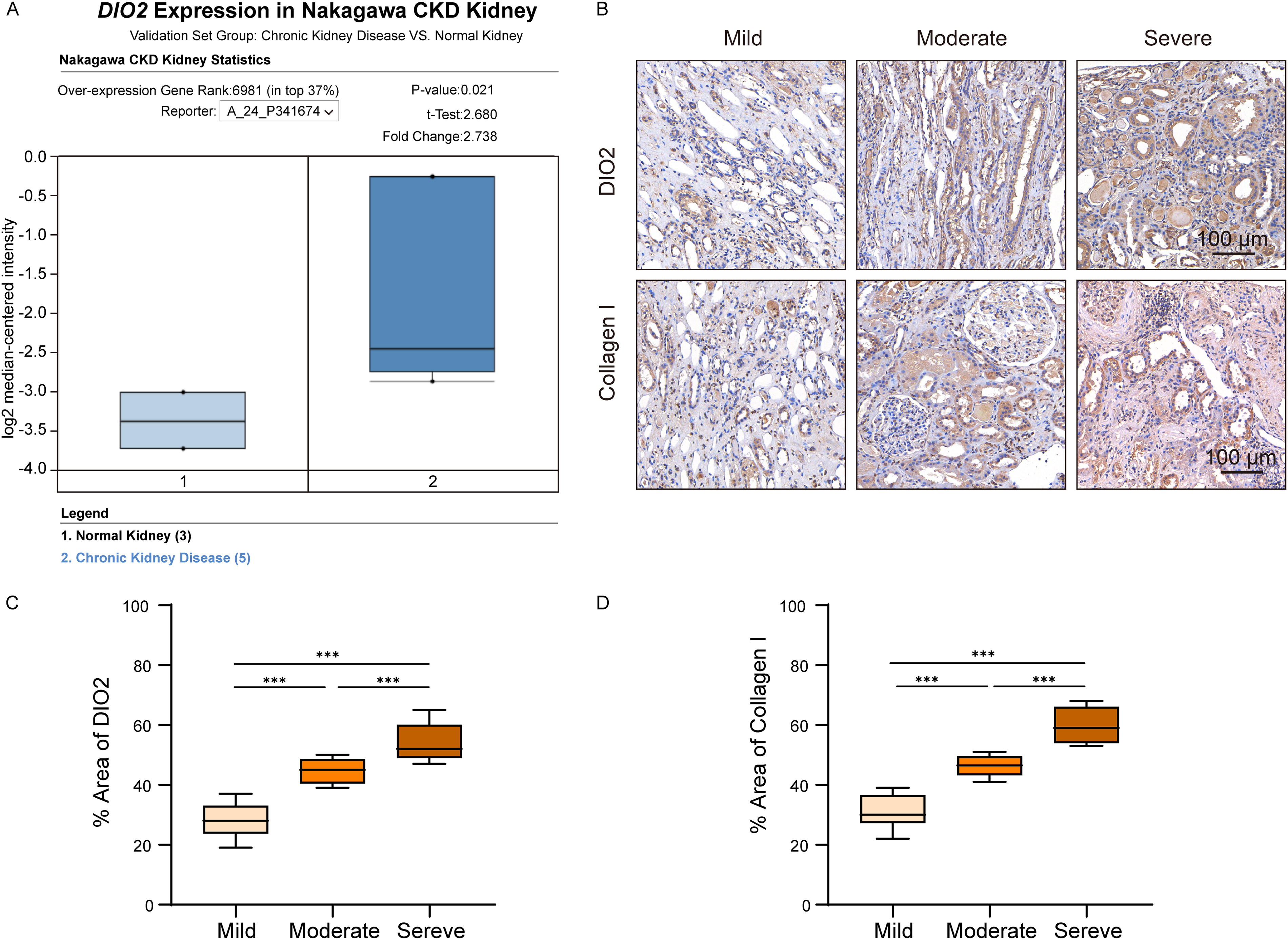
Significant upregulation of DIO2 in patients with CKD and its expression changes across different fibrosis severities.
IHC analysis of renal biopsy samples from patients with CKD examined the correlation between DIO2 expression and fibrosis severity (Fig. 6B). Based on pathological grading criteria, the samples were categorized into mild (n = 8), moderate (n = 6), and severe (n = 7) fibrosis groups. IHC staining revealed a progressive increase in DIO2 expression with fibrosis severity, with notably strong staining observed in the severe fibrosis group. To quantify the changes in DIO2 expression, the proportion of DIO2-positive staining areas was analyzed across the three groups (Fig. 6C). Statistical analysis showed significant differences in DIO2 expression among the mild, moderate, and severe fibrosis groups.
Additionally, to confirm whether changes in DIO2 expression correlate with the degree of fibrosis, IHC staining of Collagen I was also performed (Fig. 6B). As a hallmark protein of fibrosis, Collagen I expression was found to increase significantly with the severity of fibrosis. Quantitative analysis showed significant differences in Collagen I-positive staining areas among the different fibrosis groups (Fig. 6D). The consistent expression patterns of DIO2 and Collagen I strongly support DIO2 as a key molecule involved in renal fibrosis pathology, and its role as a potential therapeutic target deserves further investigation.
Validation of DIO2 expression changes and its association with fibrosis in in vivo and in vitro
To further verify the expression changes of DIO2 and its association with fibrosis in in vivo and in vitro models, relevant experimental validations were performed. In the in vivo UUO mouse model, Western blot and qRT-PCR results revealed that UUO induced a marked upregulation of both protein and mRNA expression of DIO2. Treatments with DPSC and HGF-DPSC significantly suppressed this upregulation, with the HGF-DPSC group exhibiting the stronger inhibitory effect (Fig. 7A and B). IF staining further demonstrated that DIO2 was highly expressed in the UUO group and co-expressed with fibrosis markers Collagen I and α-SMA (Fig. 7C). Following treatment with DPSC and HGF-DPSC, the expression of these markers was substantially reduced, with the HGF-DPSC group showing the more pronounced improvement.
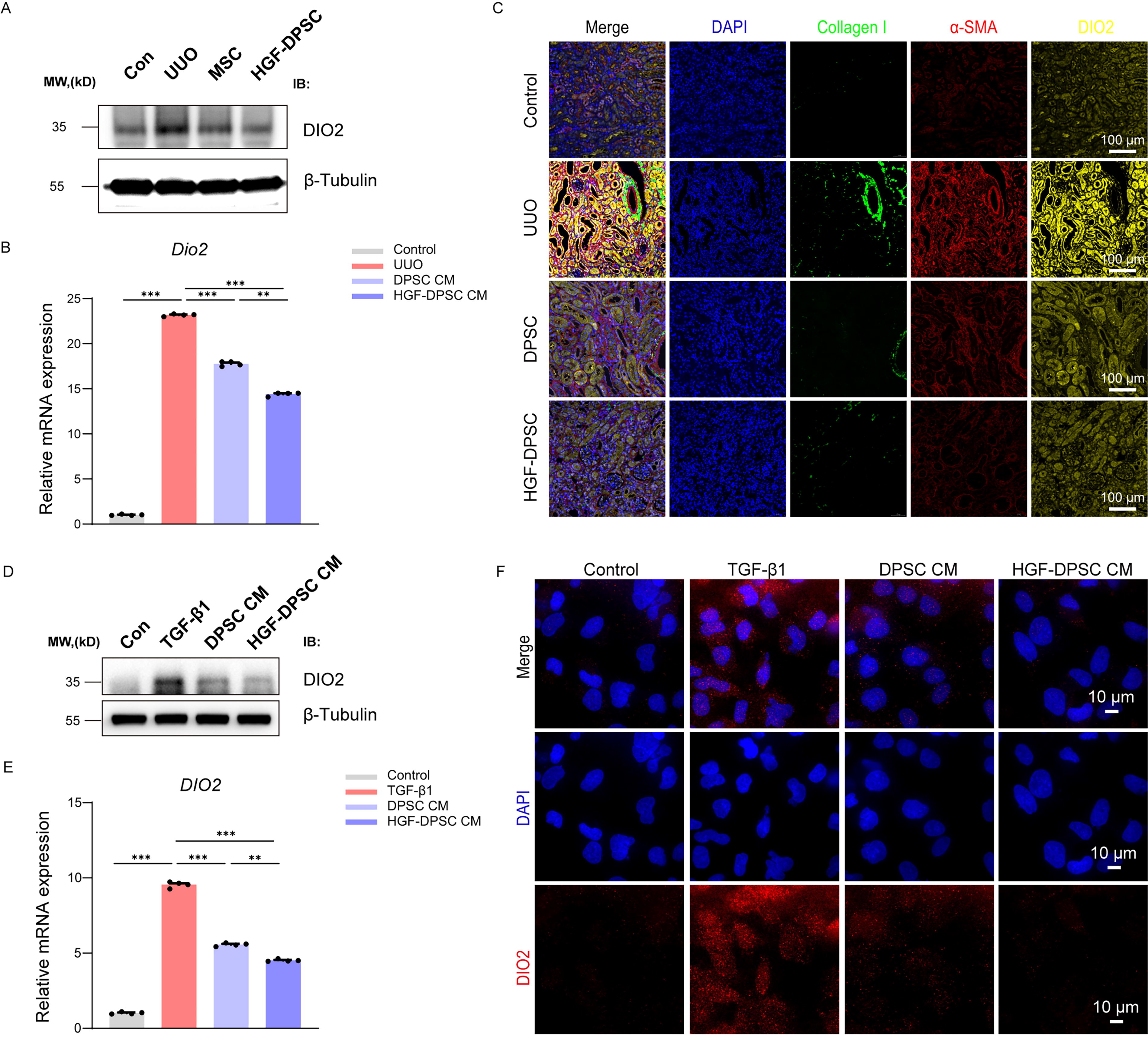
Validation of DIO2 expression changes in in vivo and in vitro. Western blot
In the in vitro TGF-β1–induced HK-2 cell fibrosis model, Western blot and qRT-PCR results similarly showed that TGF-β1 induced a significant increase in DIO2 expression, while treatments with DPSC CM and HGF-DPSC CM effectively suppressed its expression. The inhibitory effect was more pronounced in the HGF-DPSC CM group (Fig. 7D and E). IF analysis confirmed that the elevated DIO2 signal induced by TGF-β1 was markedly reduced by DPSC CM and HGF-DPSC CM treatments (Fig. 7F).
Collectively, these in vivo and in vitro findings consistently demonstrate that DIO2 is significantly upregulated during the fibrotic process and that its expression can be effectively downregulated by DPSC and HGF-DPSC treatments, with the latter exhibiting the stronger effect. These findings further highlight DIO2 as a key molecule associated with renal fibrosis progression.
HGF mediates antifibrotic effects through direct interaction with DIO2 as part of the PI3K/AKT/GSK3β-DIO2 signaling axis
In this study, transcriptomic analysis revealed that Dio2 was the most significantly downregulated gene in the HGF-DPSC treatment group. To the best of our knowledge, no prior studies have reported the role of DIO2 in renal fibrosis. This finding suggests that DIO2 may play a role in the fibrotic microenvironment. Based on this result, we conducted a preliminary exploration of DIO2 through gene network analysis, molecular docking, and cellular experiments, aiming to propose a reasonable hypothesis and provide a foundation for future studies.
GeneMANIA network analysis indicated that HGF interacts with multiple fibrosis-related signaling molecules, such as VEGFA, FGF2, and PIK3R1, forming a complex molecular interaction network (Fig. 8A). Among these associated factors, DIO2 was predicted to be a potentially significant molecule related to HGF, providing a clue for further investigation. Subsequently, we performed molecular docking analysis to predict the binding mode between HGF and DIO2 (Fig. 8B). The results showed a high binding affinity between HGF and DIO2, with a binding free energy of −55.68 kcal/mol, a docking score of −288.19, and a confidence score of 0.9407. Structural analysis of the binding sites suggested that HGF and DIO2 might form a stable complex through hydrogen bonding and hydrophobic interactions. This computational prediction provided strong evidence for a potential direct interaction between HGF and DIO2.
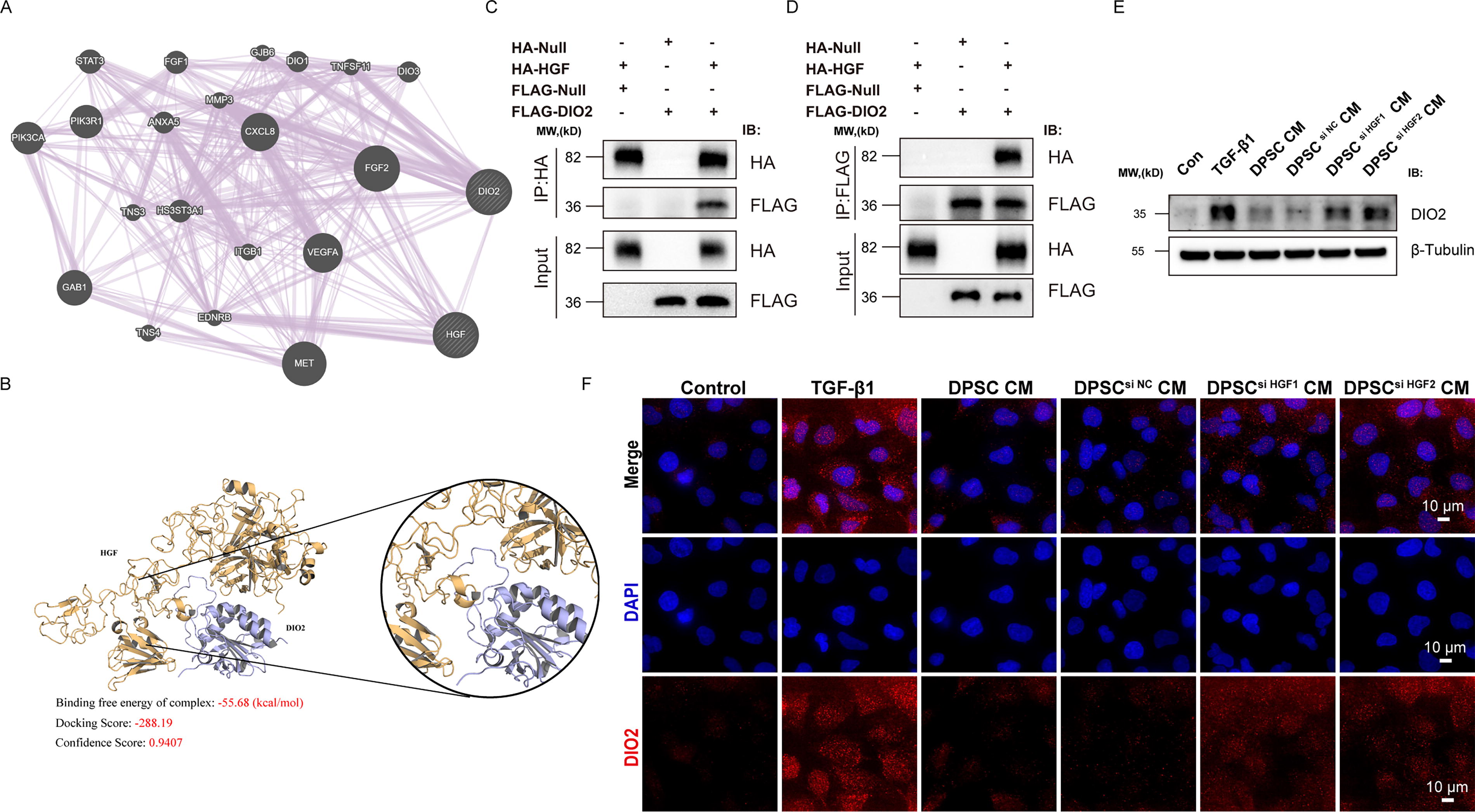
Direct HGF
To validate this predicted interaction, we conducted co-immunoprecipitation (Co-IP) experiments using HA-tagged HGF and FLAG-tagged DIO2. When immunoprecipitating HA-HGF, we detected co-precipitation of FLAG-DIO2 (Fig. 8C). Similarly, in the reverse experiment, immunoprecipitation of FLAG-DIO2 pulled down HA-HGF (Fig. 8D), confirming direct protein–protein interaction between HGF and DIO2.
We further examined DIO2 expression levels using Western blot and IF to preliminarily explore its potential regulatory mechanisms. Western blot analysis (Fig. 8E) showed that DIO2 expression was significantly upregulated in the TGF-β1–induced HK-2 fibrosis model, while treatment with DPSC CM or DPSCsi NC CM markedly reduced DIO2 expression. In contrast, the use of DPSC CM prepared from HGF-knockdown DPSC (DPSC si HGF CM) resulted in a significantly diminished ability to suppress DIO2 expression, further confirming the critical role of HGF in regulating DIO2. These findings suggest that HGF may play a critical role in regulating DIO2 expression. IF analysis further validated the changes in DIO2 expression (Fig. 8F). The results showed that DIO2 exhibited significantly increased red fluorescence intensity in TGF-β1-induced HK-2 cells. However, treatment with DPSC CM or DPSCsi NC CM significantly reduced the red fluorescence intensity. In contrast, cells treated with DPSC si HGF CM still displayed relatively high red fluorescence intensity for DIO2. These results infer that HGF directly interacts with and regulates DIO2 as part of the PI3K/AKT/GSK3β-DIO2 signaling axis, playing a crucial role in alleviating renal fibrosis. Our findings reveal a previously unrecognized molecular mechanism underlying the therapeutic effects of HGF-DPSC in renal fibrosis.
DISCUSSION
This study systematically investigated the therapeutic potential and underlying mechanisms of DPSC and their HGF-modified counterpart (HGF-DPSC) in renal fibrosis using in vivo and in vitro models combined with transcriptomic analysis. HGF-DPSC effectively ameliorated renal fibrosis pathology, likely through enhanced paracrine function of HGF, modulation of the renal fibrotic microenvironment, and suppression of aberrant activation of fibrosis-related signaling pathways. These findings establish a foundation for the clinical translation of HGF-DPSC and provide novel insights into the molecular mechanisms underlying renal fibrosis.
Stem cell therapy has emerged as a promising strategy for treating tissue fibrosis. Various sources of MSC have demonstrated significant therapeutic efficacy in different organ fibrosis models. Bone marrow-derived MSCs (BM-MSCs) have been proven effective in skin fibrosis/systemic sclerosis models through paracrine actions and immunomodulatory functions. 54 Adipose-derived MSCs (AD-MSCs) have shown unique advantages in pulmonary fibrosis, with research demonstrating that AD-MSCs effectively mitigate bleomycin-induced pulmonary fibrosis through PGE2-dependent inhibition of lung fibroblast activation. 55 UCMSCs have displayed robust therapeutic potential in liver fibrosis models, where investigations have discovered that UCMSCs can directly modulate MMPs and suppress hepatic stellate cell activation. 56 DPSCs, owing to their unique neural crest origin, have exhibited superior reparative capacity compared to other MSCs in cardiac fibrosis. 57 Despite extensive investigation of various MSC sources individually, direct comparative studies of DPSC and UCMSC in renal fibrosis models are notably absent from the literature.
In this study, our in vitro observations revealed superior paracrine effects of DPSC-conditioned medium compared to UCMSC-derived counterparts. Moreover, we explored the immunomodulatory ability of DPSC and UCMSC in our previous experiments, and the results (unpublished data) demonstrated that DPSCs exhibited significantly higher regulatory T cell (Treg) promotion rates (209.7 ± 11.6% vs. 130.7 ± 25.7%), and Th1 cells (66.5 ± 8.1% vs. 52.4 ± 4.8%) and Th17 cells (72.3 ± 9.6% vs. 54.6 ± 9.2%) inhibition rates. This enhanced capacity to shift immune homeostasis toward an anti-inflammatory profile makes DPSC particularly suitable for treating CKD, wherein persistent inflammation drives fibrosis progression.
HGF is a multifunctional paracrine factor and was identified as a key mediator of the antifibrotic effects of DPSC in this study. Knockdown of HGF expression significantly impaired the ability of DPSC to suppress renal fibrosis markers, such as Collagen I and α-SMA, and to regulate fibrosis-related signaling pathways, further underscoring the critical role of HGF. Previous studies have shown that HGF, by activating its receptor c-Met, 52 can effectively inhibit fibroblast activation and excessive ECM deposition while promoting the repair and regeneration of renal tubular epithelial cells. 58,59 Consistently, HGF-DPSC demonstrated superior antifibrotic effects compared to unmodified DPSC, particularly in regulating the renal fibrotic microenvironment. Transcriptomic analysis further revealed that HGF-DPSC downregulated ECM-related and pro-inflammatory genes, thereby reshaping fibrosis-associated gene networks and signaling pathways. These results highlight the importance of HGF modification in enhancing the antifibrotic efficacy of DPSC.
The PI3K/AKT/GSK3β signaling pathway was identified as a critical target of HGF-DPSC in renal fibrosis. This pathway is widely involved in ECM over-deposition and aberrant expression of fibrosis markers. 60 –62 In the UUO model, this pathway was significantly activated, whereas HGF-DPSC treatment markedly reduced the phosphorylation levels of PI3K, AKT, and GSK3β and suppressed abnormal nuclear translocation of β-catenin. These findings suggest that HGF-DPSC can achieve renal fibrosis amelioration through multitarget regulation of this pathway, providing a new therapeutic strategy targeting PI3K/AKT/GSK3β signaling.
This study identified DIO2 as a novel and critical mediator in renal fibrosis. Transcriptomic analysis revealed that Dio2 was the most significantly downregulated gene following HGF-DPSC treatment. Although the role of DIO2 in renal fibrosis has not been previously reported, our comprehensive analysis of human CKD samples confirmed a strong positive correlation between DIO2 expression and fibrosis severity, consistent with findings in idiopathic pulmonary fibrosis. 63 Most significantly, we experimentally validated direct protein–protein interaction between HGF and DIO2 through Co-IP, confirming our computational predictions from molecular docking. These findings not only identify DIO2 as a novel biomarker for renal fibrosis severity but also suggest that specifically targeting DIO2 could represent a promising therapeutic strategy for treating renal fibrosis. Future studies should explore the development of DIO2 inhibitors as potential therapeutics for CKD and investigate whether DIO2 plays similar roles in fibrotic conditions affecting other organs. At present, the specific function and mechanism of DIO2 in fibrosis pathology remain unclear. In future studies, we plan to conduct functional experiments, such as DIO2 knockout or overexpression, to validate its role in fibrosis. These questions will be explored in greater depth in subsequent work to elucidate the precise role and molecular mechanisms of DIO2 in renal fibrosis.
It is worth noting that MSC-based therapeutics have achieved significant progress in clinical development. Recently, RYONCIL®64 from Mesoblast Ltd. and PLEB001 65 from Platinumlife Biotechnology Co., Ltd. were approved by the Food and Drug Administration and the National Medical Products Administration successively for the treatment of steroid-refractory acute graft-versus-host disease. So far, 16 MSC drugs have been approved worldwide. These milestones highlight the clinical potential of MSC-based therapies and provide critical inspiration for MSC gene modification technologies, such as HGF-DPSC. Currently, there are no approved MSCs for the treatment of kidney diseases. The multitarget regulatory capabilities of HGF-DPSC demonstrated in this study establish a foundation for their clinical application in CKD and other renal fibrosis-related diseases.
Despite the comprehensive exploration of HGF-DPSC in renal fibrosis, the long-term therapeutic efficacy and safety of HGF-DPSC require further evaluation in preclinical and clinical settings. Future studies will employ high-throughput multi-omics 66 to dissect the dynamic changes of cellular populations and signaling pathways in the renal fibrotic microenvironment. It is also worth exploring the spatial distribution 67 of HGF-modified DPSC during the treatment of CKD. Further investigations will also focus on validating the molecular mechanisms of HGF-DPSC and assessing their long-term therapeutic potential and safety in CKD and other fibrotic diseases.
In conclusion, our study demonstrates that HGF-modified DPSCs significantly alleviate renal fibrosis through multiple mechanisms. We identified DIO2 as a novel fibrosis-associated gene regulated by HGF and elucidated the role of the PI3K/AKT/GSK3β pathway in mediating these antifibrotic effects. The direct interaction between HGF and DIO2 revealed by our Co-IP experiments represents a previously unrecognized molecular mechanism with potential therapeutic implications. These findings not only advance our understanding of renal fibrosis pathogenesis but also highlight the promising clinical application of genetically modified DPSCs as a cell-based therapy for CKD.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to China Medical Management Consulting and Beijing SH Biotechnology for gifting their clinical-grade UCMSC and DPSC.
DATA AVAILABILITY
All data generated or analyzed during this study are included in this published article and its Supplementary Figures. The data supporting the findings of this study and datasets used in this study are available from the corresponding author upon reasonable request. Transcriptomic data have been deposited in the China National Center for Bioinformation under project number PRJCA036337. The authors will publish the data after the article is published.
ETHICS APPROVAL STATEMENT
Clinical-grade UCMSC was obtained from China Medical Management Consulting Co., Ltd. (Beijing), the approval number is 2023-002-001. Clinical-grade UCMSC was obtained from Beijing SH Biotechnology, the approval number is CMUSH-IRB-KJ-YJ-2022-13. All procedures that involved animals were approved by the IACUC of the Laboratory Animal Center of the Academy of Military Medical Sciences (IACUC-DWZX-2023-557). The patient’s tissue slice passed the ethical review, the approval number is K2025134(EZ).
AUTHORS’ CONTRIBUTIONS
J.S.: Performed the experiments, collected data, and prepared the article. W.X.: Contributed renal biopsy specimens and performed partial IHC analyses. N.T., H.D., Z.H., and L.W.: Data analysis and interpretation. C.-T.W.: Administrative support. H.W.: Conception, design, and financial support. All the authors read and approved the final article.
AUTHOR DISCLOSURE
The authors have no potential conflicts of interest to declare.
FUNDING INFORMATION
No funding was received for this article.
SUPPLEMENTARY MATERIAL
Supplementary Figures
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.