
Abstract
Proteinase 3 (PR3), a neutrophil granule serine protease, is the major autoantigen for autoantibodies in the systemic vasculitic disease, Wegener's granulomatosis. It is also found to be involved in various inflammatory diseases including Crohn's disease, rheumatoid arthritis, cystic fibrosis, and gingivitis. However, there is no high quality antibody available to detect endogenous PR3 in biological samples such as plasma and tissue. Several commercial anti-PR3 monoclonal antibodies (MAbs) were obtained by using HMC-1/PR3 cell granule extracts as the antigen, but the resulting antibodies could not be applied for immunoblotting or other immunological methods. Therefore, we produced human recombinant PR3 in Escherichia coli and developed several MAbs that are highly sensitive and can be used for immunoblotting, FACS analysis, and immunofluorescent staining. The PR3 MAbs recognized both rhPR3 and human plasma–derived neutrophil PR3 in reducing and non-reducing conditions at low nanogram levels. In addition, new MAbs detect endogenous PR3 from normal human plasma and urine with high specificity. The new anti-PR3 MAbs will be an essential tool for investigating the role of PR3 in inflammatory and autoimmune diseases.
Introduction
Besides PR3 activity on the cell surface and in the extracellular space, the protease also has intracellular affects. For example in endothelial cells, it mimics caspase activity by cleaving NF-κB and inducing sustained JNK activation.(2) PR3 can also cleave and inactivate the major cell-cycle inhibitor p21 Waf1/Cip1/Sdi1 .(11) High levels of PR3 and p21 cleavage products were found in intestinal biopsies from patients with Crohn's disease or ulcerative colitis.(12) It also appears that the role of PR3 in autoimmune disease extends beyond these functions. The frequency of the membrane bound PR3 is increased in patients with anti-neutrophil cytoplasmic autoantibody-associated vasculitis as well as in patients with rheumatoid arthritis; therefore PR3 is considered to be a risk factor in these diseases.(13)
There are several reports correlating PR3 to other diseases. Gene expression profiles of peripheral neutrophils and monocytes from patients with anti-neutrophil cytoplasmic autoantibody-related kidney diseases showed increased levels of PR3 transcripts, and the expression correlated with disease activity and with glomerulonephritis.(14,15) In patients with cystic fibrosis (CF), increased levels of PR3 mRNA have been reported in circulating monocytes upon exacerbation of pulmonary disease.(16) Surfactant protein D is an innate host defense molecule present in the lung of CF-affected patients and is a target for PR3.(17) One hypothesis is that impaired host defense against bacterial colonization in patients with CF may be due to increased proteolysis of surfactant protein D by PR3, thereby increasing the incidence of active lung infection. In patients with gingivitis and periodontitis, endogenous PR3 is expressed in oral epithelial cells, and anti-neutrophil cytoplasmic autoantibodies are found in the patient's serum.(18)
Although the high association between PR3 expression and these diverse diseases has been frequently demonstrated, antibodies with specificity and sensitivity for detection of endogenous PR3 in biological samples, such as blood, or clinical diagnostic methods have not been produced. In fact, several commercial anti-PR3 monoclonal antibodies obtained by using HMC-1/PR3 cell granule extracts as antigen are currently marketed for research purposes. However, these antibodies only partially recognize the neutrophil PR3 or recombinant PR3 and with a limited range of detection. Therefore, these antibodies are not appropriate for the detection of very small amounts of PR3 in biological samples by immunological methods such as immunoblotting, FACS analysis, and immunohistochemistry.
In the present study, we expressed recombinant huPR3 (rhPR3) in E. coli for use as antigens to produce new anti-PR3 MAbs that could be sensitive enough to detect intrinsic PR3 in human blood and urine and to generate diagnostic methods of immunoblotting, FACS analysis, and immunohistochemistry. The anti-PR3 MAbs and detection methods obtained from this study will be useful tools for investigating the role of PR3 in inflammatory autoimmune diseases.
Materials and Methods
Construction of recombinant human PR3
Human PR3 cDNA (GenBank accession no. NM 002777) was cloned from the THP-1 cell line using RT-PCR with forward primer 5′-AGAGGAGCTTGATCGT GGGTG-3′ and reverse primer 5′-GAGCTGCTTCTGTCCAAAGATC-3′. After TA cloning with TOPO vectors (Invitrogen, Carlsbad, CA), the sequence was confirmed by DNA sequencing. A pair of primers was designed, which has an EcoRI site at the 5 end and an XbaI site at the 3 end, in order to construct an E. coli expression plasmid vector. The PCR product of mature human PR3 sequence was digested with EcoRI and XbaI and then inserted into the pPROEx HTa plasmid (Invitrogen) to generate huPR3/pPROEx HTa. The constructed huPR3/pPROEx HTa plasmid was transformed into DH5α competent cell for protein expression.
Recombinant protein expression and purification
Recombinant huPR3 protein was expressed in E. coli DH5α cells, which were cultured in LB broth containing 100 μg/mL ampicillin. The cultures were incubated at 37°C with constant shaking until the optical density at 600 nm reached 0.5∼0.6. Human recombinant PR3 protein was induced by adding IPTG (0.6 mM) and further cultured at 37°C with constant shaking for 3 h. The bacteria were harvested by centrifugation at 9500 g for 20 min.
The cell pellet was resuspended in 25 mL of buffer A (20 mM Tris–HCl [pH 8.0], 8 M urea) per gram of cells. The cells were disrupted by sonication and solubilized at room temperature (RT) with constant shaking for 1 h. The soluble proteins were harvested by centrifuging at 9500 g for 20 min at RT. The supernatant was dialyzed against buffer B (20 mM Tris–HCl [pH 8.0], filtered) using dialysis bags with a 6–8 kDa molecular weight cut-off. The dialyzed human recombinant PR3 protein was centrifuged again at 4°C for 20 min in order to remove any aggregated proteins.
In the pPROEx HTa expression vector, the human PR3 sequence is followed at its N-terminus by the six histidines and TEV protease cleavage sites for affinity purification using cobalt chelate affinity chromatography. The supernatant was loaded onto a cobalt-charged Talon column (Clontech Laboratories, Mountain View, CA), and the column was washed with 50 column volumes of buffer C (20 mM Tris-HCl [pH 8.0], 300 mM NaCl) containing 0.1% of TritonX-114 for endotoxin removal.(19) The His-tagged huPR3 protein was eluted using buffer D (20 mM Tris–HCl [pH 8.0], 300 mM NaCl, 300 mM imidazole). The fractions containing the protein of interest were dialyzed against buffer B (20 mM Tris-HCl [pH 8.0], filtered) using the same dialysis bags. The N-terminus six histidines were removed by TEV protease.
The protein was further purified by Superdex 75 HR 10/30 gel filtration (GE Healthcare Biosciences, Piscataway, NJ) using a FPLC system with buffer B (20 mM Tris-HCl [pH 8.0], filtered). The recombinant protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and identified by mass spectrometry (data not shown).
Generation, selection, and purification of anti-PR3 MAbs
Pathogen-free female BALB/c mice (6 weeks of age) were enrolled in this study and maintained in a room at 22°C under a 12 h light/dark cycle. The mice were located in the Medical Biological Science Institute at Konkuk University and all animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Konkuk University.
Purified recombinant huPR3 proteins (rhuPR3) were used to immunize female BALB/c mice, 6 weeks of age, by subcutaneous injections. The mixture of Freund's complete adjuvant (Sigma-Aldrich, St. Louis, MO) and rhuPR3 was used for the two immunizations with a 2 week interval. The mixture of Freund's incomplete adjuvant and rhuPR3 was used for the third immunization. Mice were bled 10 days after each boost. The titer of mouse serum was determined by direct enzyme-linked immunosorbent assay (ELISA). The last boost of antigen was injected 3 days before a fusion.
FO myeloma cell line was fused with splenocytes from immunized mice according to standard protocols. The hybridoma cells of positive wells were cloned by limiting dilution in aminopterin-free selection Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum. The single clones were screened by direct ELISA, dot blotting, and Western blotting for their specificity to rhuPR3. A single hybridoma clone was expanded for further purification. Anti-huPR3 MAb IgG was purified from hybridoma cell culture supernatants using protein G agarose (KPL, Gaithersburg, MD).
Subclass determination
To determine the subclass of each MAb to PR3, direct PR3 ELISA was performed as described below. Bound anti-PR3 MAbs were incubated with subclass-specific antibodies, rat anti-mouse IgG1, IgG2a MAb HRP-labeled conjugate (GeneTex, San Antonio, TX), or goat anti-mouse total IgG Ab HRP-labeled conjugate (Millipore, Temecula, CA). The optical density (OD) values were detected in a Spectramax M190 reader at 450 nm (Molecular Devices, Sunnyvale, CA).
Direct ELISA
For direct ELISA, a Maxisorp plate (SPL) was coated with 1μg/mL of rhuPR3 and incubated overnight at 4°C. Non-specific protein binding sites were blocked with 3% BSA in PBS for 2 h at RT. Plates were washed with PBS containing 0.1% Tween-20 (PBS-T). Sera from immunized mice, serially diluted in PBS, and cell culture supernatants were added and incubated at RT for 2 h in the coated plates. After washing with PBS-T, a 1:10,000 dilution horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Millipore) was added for 40 min at RT. After washing, 3,3′,5,5′-tetramethylbenzidine base substrate (Millipore) was added. The color reaction was stopped with 0.1 mL of 1 M H2SO4 to each well, and the plates were examined at 450 nm in an ELISA reader.
Western blot analysis
Recombinant huPR3 and human neutrophil proteinase 3 (Athens Research & Technology, Athens, GA) samples mixed with Laemmli sample buffer were boiled for 10 min and loaded on the 10% SDS-PAGE. Samples were electrotransferred onto a nitrocellulose membrane and blocked with 5% skim milk in PBS-T. Membrane was probed with the protein G purified antibodies as primary antibodies and the bound antibodies were detected by horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Millipore) at dilution 1:10,000 followed by ECL substrate solutions (GE Healthcare Biosciences).
Immunofluorescent staining
For indirect immunofluorescent staining, THP-1 cells grown on Permanox Labtek chambers (Nunc, Rochester, NY) were fixed with 100% acetone at RT for 20 min and then washed three times at RT. Cells were blocked with 3% BSA containing PBS for 1 h at RT and washed three times at RT. Then cells were incubated with mouse IgG1 control (eBioscience, San Diego, CA), biotinylated anti-rhuPR3 monoclonal IgG clone 32, and commercial mouse anti-PR3 monoclonal antibody (Biomeda, Foster City, CA). After extensive washing with PBS, cells were further incubated with secondary antibody (Streptavidin-FITC or anti-mouse-FITC, 1:1000; Pierce, Rockford, IL) in PBS at RT for 1 h, and then incubated with DAPI (1:5000) in PBS at RT for 10 s. The samples were mounted with 5 μL medium, dried in the dark, and observed by fluorescence microscopy.
Human plasma and urine samples
Whole blood sample from normal subjects was obtained in EDTA tubes. The whole blood was kept at 4°C and centrifuged within 3 h. The plasma was carefully aspirated with a Pasteur pipette and stored at −20°C until assayed. Human urine samples were collected in normal subjects and centrifuged prior to using for Western blot analysis.
Results
Expression and purification of recombinant human PR3 as antigen
huPR3/pPROEx HTa plasmid vector was used to produce recombinant huPR3 protein, which was first purified with a cobalt chelate affinity chromatography. The eluted recombinant huPR3 protein fractions were separated on 10% SDS-PAGE and visualized by Coomassie blue staining (Fig. 1A). The expected size of fusion rhPR3 protein, including His 6 -tag and TEV protease cleavage site, was about 34 kDa. However, there were two major bands with molecular weights of about 34 kDa and 24 kDa (Fig. 1A). Therefore, we examined both 34 and 24 kDa bands with mass spectrometry analysis. The mass spectrometry data showed that both molecular size bands were identified as huPR3 protein (data not shown). In order to remove the N-terminus His 6 -tag fusion part, rhuPR3 protein was cleaved with TEV protease, resulting in both 34 kDa and 24 kDa decreased molecular size as 30 kDa and 20 kDa in Coomassie blue staining (Fig. 1B).

Preparation of recombinant huPR3 proteins as antigen. (
Production of MAbs against recombinant huPR3 protein
Five Balb/c mice were immunized with the purified rhuPR3 protein and successful fusions were achieved using splenocytes from two immunized Balb/c mice, as described in the Materials and Methods section. For screening of antibody producing hybridoma clones, recombinant huPR3 was used as antigens with direct ELISA. We screened 38 hybridoma clones with direct ELISA and found 10 positive hybridoma clones (Fig. 2). Six hybridoma clones (4, 19, 27, 32, 36, and 38) survived while we further expanded them to obtain a single clone. Hybridoma cell culture supernatants were obtained and their immunoglobulin subclasses were determined. All of them were identified as IgG1 subclass (Table 1).
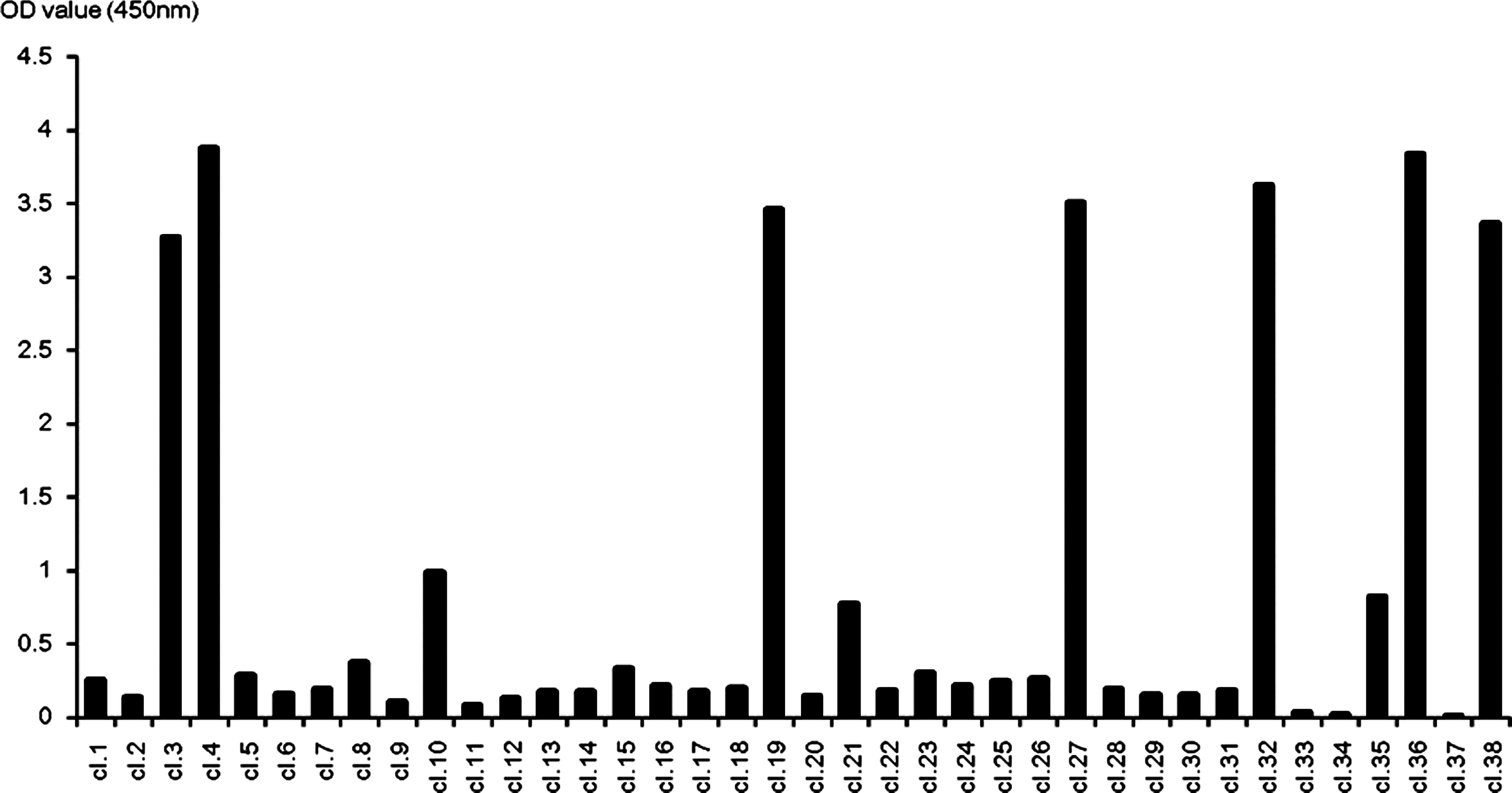
Screening the hybridoma cells of anti-human PR3. Direct ELISA revealed the supernatant of 10 hybridoma clones reacting with rhuPR3 from 38 different clones. The supernatant of seven clones (3, 4, 19, 27, 32, 36, and 38) showed significant OD value (>3.00), and those clones were continued for limiting dilution and further characterization.
Immunoblot with selected anti-rhuPR3 MAbs
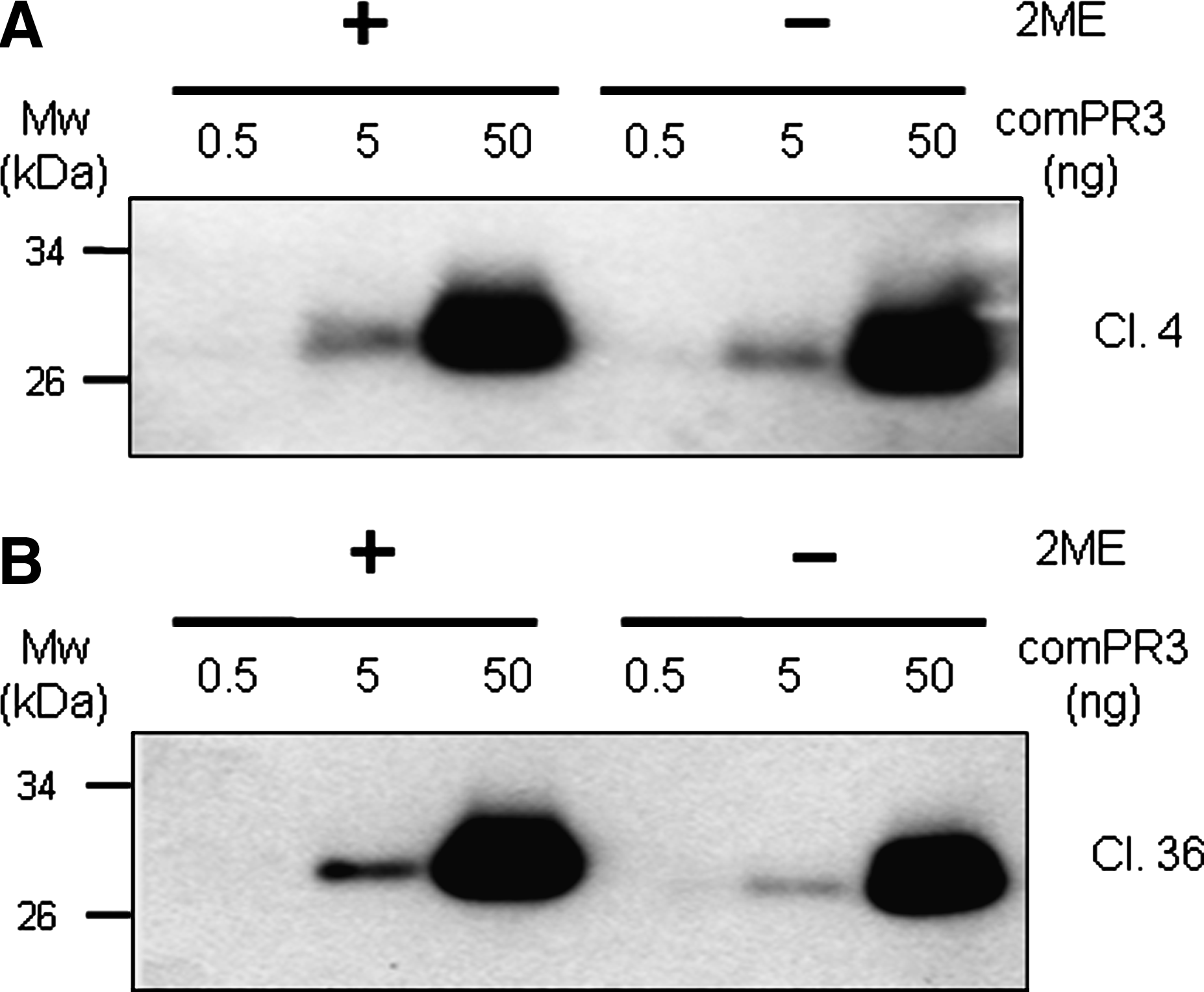
Six hybridoma clones produced MAbs that were able to react with rhuPR3 protein under the reducing or non-reducing conditions of Western blotting, whereas clone 19 MAb was not able to recognize the commercial PR3 (Fig. 3B). The commercial PR3 is a monomeric form and is enzymatically active whereas rhuPR3 from E. coli is mostly a multimeric form with an intra-disulfide bond, which was observed by silver staining (data not shown). As shown in Figure 3A–C, clones 4 and 27 detected both monomer and multimer forms, except for clone 19. Interestingly clone 32, 36, and 38 poorly recognized the non-reduced multimeric rhuPR3 form (Fig. 3D–F).

Western blot analysis for specificity to rhuPR3 and neutrophil PR3 in reduced or non-reduced condition. Western blots were performed with the supernatants of anti-rhuPR3 antibody-producing hybridoma clones. The purified rhuPR3 and neutrophil PR3 (commercial PR3) were loaded in each lane (50 ng), as indicated at the top of the figures (reducing or non-reducing conditions), and transferred to membrane. The Western blot membranes were probed with the supernatant of hybridomas clones (4(
Immunoblotting was performed with commercial PR3 (human neutrophil PR3) in order to determine the detection limit of anti-rhuPR3 MAbs (Fig. 4). We used two MAb clones, 4 and 36, because these two clones exhibited a different pattern of PR3 protein recognition (Fig. 3). For example, MAb clone 4 sufficiently recognized both recombinant huPR3 and commercial PR3 (Fig. 3A) whereas MAb clone 36 poorly detected recombinant huPR3 (Fig. 3E). However, both MAb clones are able to detect a few nanograms of human neutrophil PR3 under both reducing and non-reducing conditions (Fig. 4).
Detection limit of commercial PR3 with two different MAbs against human PR3. Various concentrations of commercial PR3 were subjected to SDS-PAGE gel and transferred to membrane. The membrane was probed with MAb (clones 4 and 36 with 1 μg/mL) in both reduced and non-reduced conditions. Both MAbs (clones 4(
Immunofluorescent staining of native form of PR3 in THP-1 cells
The detection ability of MAbs to endogenous PR3 was further investigated by immunofluorescent staining of cells (Fig. 5). We examined all six MAbs obtained from the hybridoma clones (data not shown). Anti-hPR3 MAb clone 32 strongly reacted with endogenously synthesized PR3 proteins in THP-1 cells when used for immunofluorescent staining (Fig. 5A, right panel) whereas the negative control mouse IgG showed no staining (Fig. 5A, left panel).
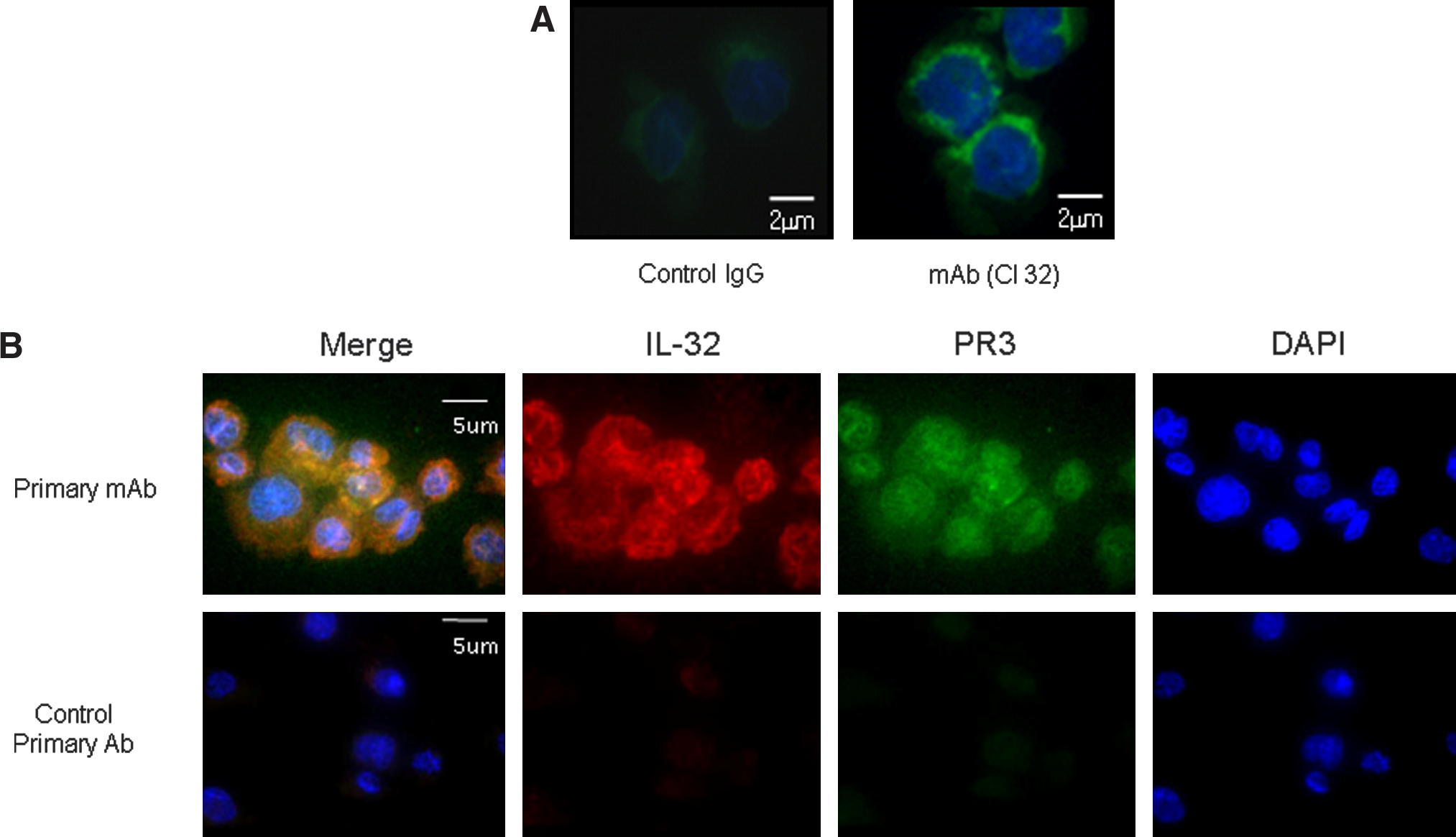
Immunofluorescent staining of endogenous PR3 and co-localization of PR3 and IL-32 in THP-1 cells. (
We further analyzed MAb clone 32 to determine if it can be used for double immunofluorescent staining. IL-32 is a proinflammatory cytokine that interacts with PR3 with a high affinity (few tens nanomole range). It is known that IL-32 is expressed in THP-1 cells (unpublished data). We used MAb clone 32 and affinity purified goat anti-IL-32 polyclonal antibody for double immunofluorescent staining. As shown in Figure 5B, goat anti-IL-32 polyclonal antibody stained endogenous IL-32 (red in second top panel) and MAb anti-PR3 clone 32 specifically stained endogenous PR3 (green in third top panel); DAPI staining showed the nuclei of cells in blue (fourth top panel). In order to show co-localization, overlapping of the two antibodies' stained images are illustrated in orange (Fig. 5B, first top panel). In the bottom panel of Figure 5B, the control primary mouse IgG did not stain cells, except for nuclear staining by DAPI.
Flow cytometry analysis with THP-1 and U937 cells
Two monocytic human cell lines were used to analyze expression of PR3 on the cell surface. Endogenous PR3 exists in different locations of the cell. An enzymatically active mature form of PR3 usually resides in secretory vesicles of neutrophils and monocytes whereas an immature form of PR3 exists on the cell surface of monocytes as glycosyl-phosphatidylinositol-anchored membrane-bound form. We examined all six anti-PR3 MAbs to determine whether they recognize the membrane-bound form on cell surface PR3. Unlike the two distinct patterns in the Western blot results, flow cytometry results vary in the ability of anti-PR3 MAb to recognize PR3 (Fig. 6). The best result was obtained in FACS staining with MAb clone 38. Although MAb clones 4, 19, 27, 32, and 36 detected the membrane-bound form of PR3 in only THP-1 cells, its signal was very poor compared to that of MAb clone 38. Interestingly, MAb clone 19 detected a membrane-bound form of PR3 only from THP-1 cells although the signal was weaker than that of MAb clone 38.
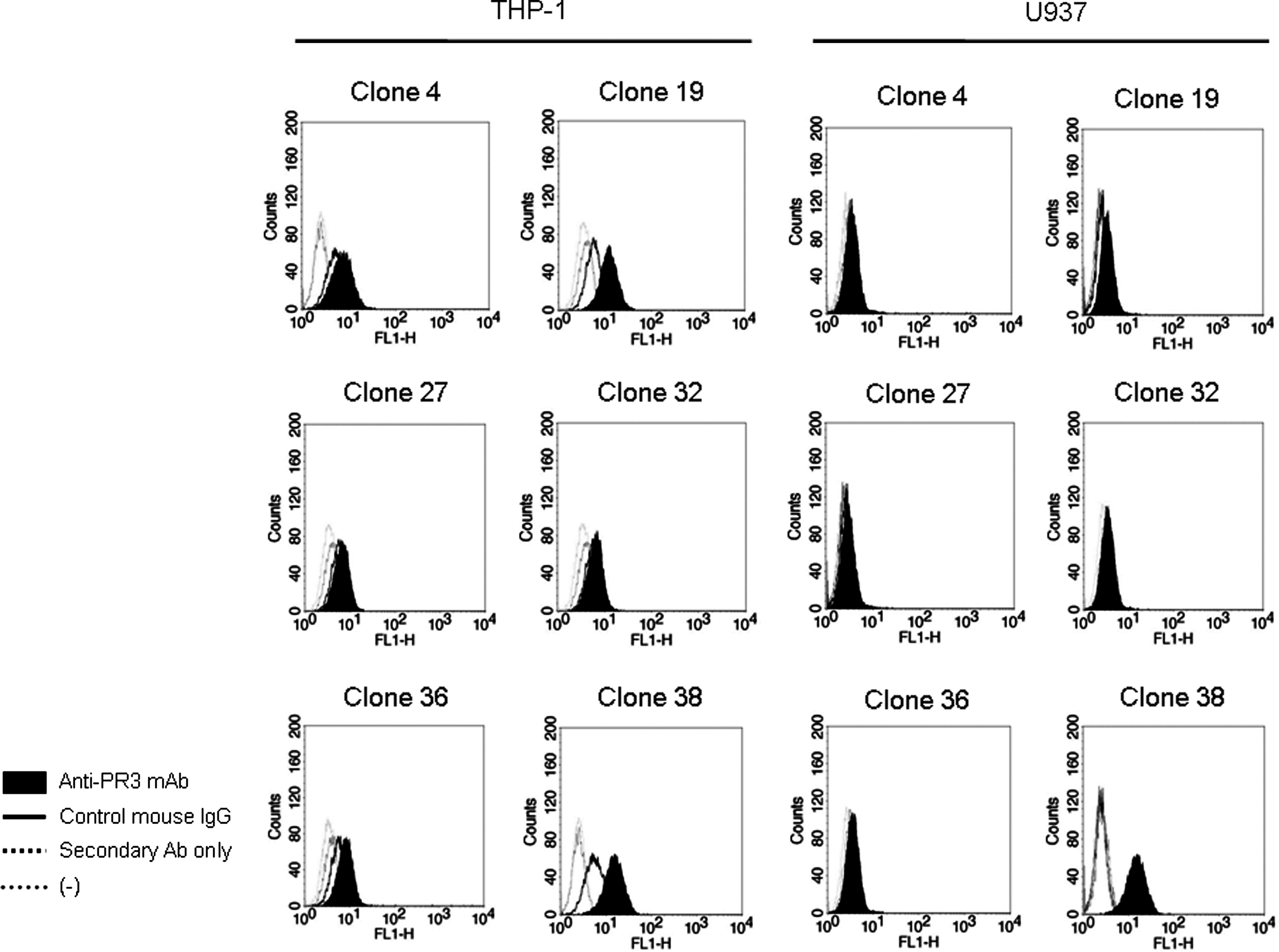
Flow cytometry of cell surface PR3 on THP-1 and U937 cells. Cell surface-bound PR3 analyzed by flow cytometry with six different anti-huPR3 MAbs. Cells were incubated with anti-PR3 MAb (0.2 μg/mL) as described in the Materials and Methods section. The membrane-bound PR3 proteins were detected in THP-1 cell U937 cell lines by using flow cytometery. Fluorescence intensity profiles were obtained with various control conditions. Control IgG is shown by bold black line. Signal revealed from secondary antibody alone is shown by black (dotted line). Non-stained cells are shown by gray dotted line. A total of 10,000 cells were analyzed in each experiment.
Detection of the endogenous PR3 in biological samples such as human plasma and urine by Western blotting
To examine the specific detection of natural PR3 in the biological samples from normal humans, Western blotting was performed using human plasma and urine. Figure 7 represents a specific and sufficient detection of natural PR3 in human plasma with protein G purified IgG of anti-huPR3 MAb clone 27. There are variations in PR3 expression in human plasma in individuals, and three distinct bands were detected in a very specific manner. The lowest molecular size band seems to be the mature form of circulating PR3 since its migration in 10% SDS-PAGE was similar to the mature form of commercial PR3 (lightest lane of Fig. 7A). The two larger molecular sizes of PR3 are complex forms with other unknown molecules in human plasma. The same MAb clone was used to detect urine PR3. As shown in Figure 7B, the MAb clone 27 sufficiently detected urine PR3 in the monomeric form. There were two high molecular bands detected in urine; however their molecular sizes slightly differ from the high molecular weight of plasma PR3. This data suggests that the new anti-huPR3 MAb could sufficiently detect the endogenous PR3 in biological samples such as human plasma and urine.
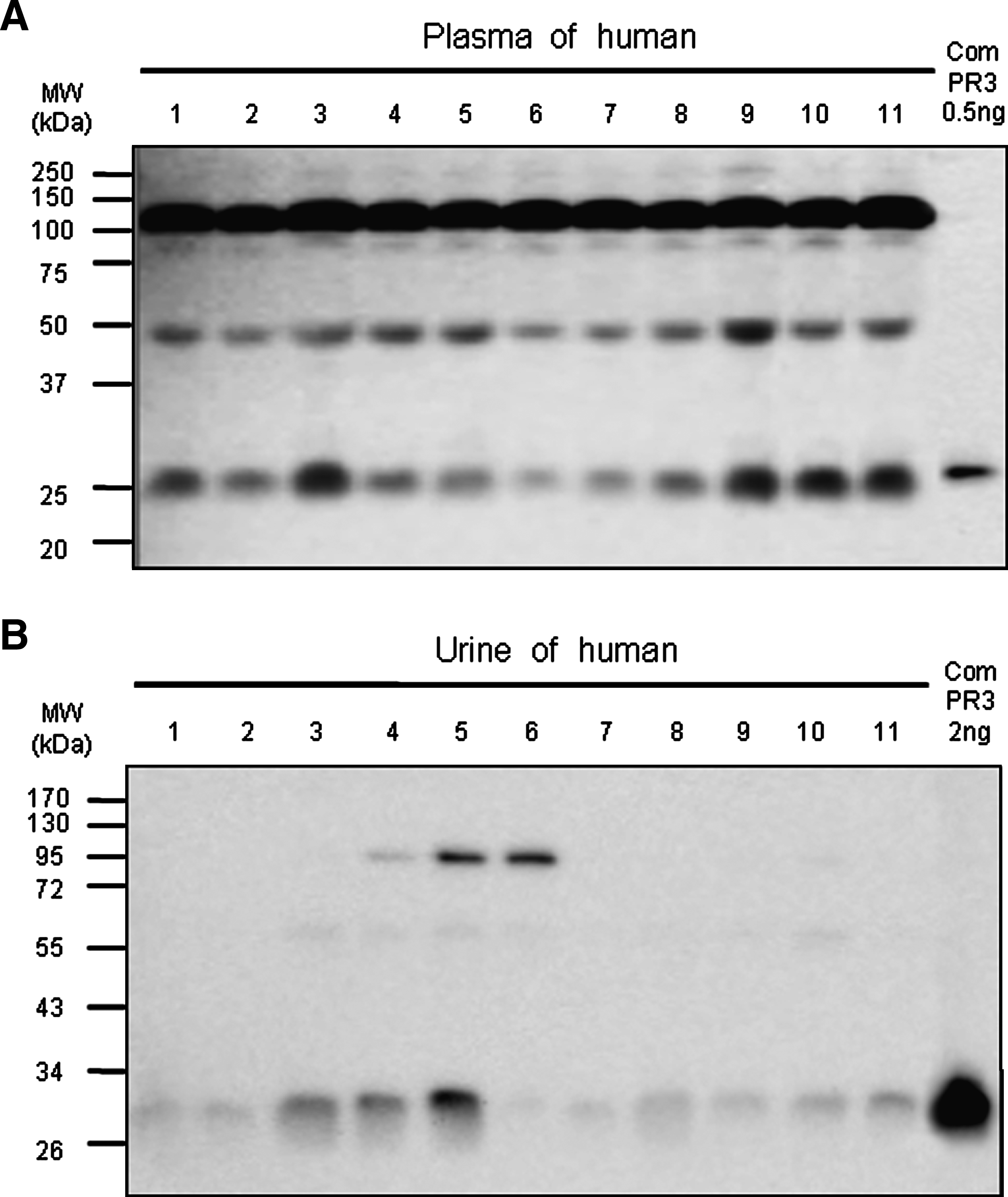
Detecting the intrinsic human PR3 in plasma and urine by Western blot analysis. (
Discussion
In the present study, we expressed human recombinant PR3 and generated six anti-human PR3 MAbs. The two distinct patterns of results found in the Western blot analysis of endogenous and recombinant PR3 were observed by anti-PR3 MAbs (Fig. 3). The first group of MAb clones 4 and 27 sufficiently recognized both recombinant and commercial PR3 whereas the MAb clone 19 detected only recombinant PR3. Although the second group of MAb clones (32, 36, and 38) recognized both recombinant and commercial PR3, the detecting signal of recombinant PR3 by those MAbs was weaker than that of the first group. The MAb clone 19 did not recognize the commercial PR3, which is only a monomer form of PR3 by 10% SDS-PAGE silver staining in both reduced and non-reduced conditions (data not shown). However, the MAb clones 32, 36, and 38 recognize the commercial PR3, which means that these MAbs detect the monomeric form of PR3 more efficiently. The MAb clones 4 and 27 detect both mono- and multi-forms of PR3. The result of Western blot analysis was distinct from the direct ELISA of commercial and recombinant PR3 antigens (Fig. 3) due to the natural variation in epitope presentation between the inherent antigen in ELISA plate and a denatured antigen in Western blot.
PR3 expression is delineated to immune cells of the granulocytic and monocytic lineage.(20) In these cell granules, PR3 is stored as a mature and enzymatic active protein.(21) The processing of PR3 protein occurs during PR3 synthesis. The prepro-form of PR3 is processed in four consecutive steps into the final mature form consisting of 222 amino acids: (1) the signal peptide is removed upon translocation of prepro-PR3 into the endoplasmic reticulum (ER), yielding a pro-form of PR3(22,23); (2) PR3 is glycosylated on both its potential N-linked glycosylation sites; (3) in the next processing step, the propeptide of PR3 (Ala-Glu) is removed while in the post-Golgi organelle.(22,23) The enzyme responsible for the removal of this propeptide is a cysteine proteinase distinct from dipeptidyl peptidase I (DPPI), which cleaves the propeptide of CatG,(24) HLE, and AZU.(25,26) After removal of the two amino acid propeptides, the substrate-binding pocket becomes accessible and PR3 becomes potentially enzymatically active(27); and (4) a seven amino acid carboxyterminal extension is removed during processing of PR3.(22,23)
We tested all six MAb anti-PR3 clones for immunofluorescent staining ability (data not shown). We obtained that the anti-PR3 MAb clone 32 strongly stained endogenously synthesized PR3 proteins in human THP-1 cells (Fig. 5A). In addition, we tested for co-localization of PR3 and IL-32 in THP-1 cells (Fig. 5B). PR3 co-localizes with endogenous IL-32, as shown in the merged image in the first top panel of Figure 5B (orange color).
The affinity of IL-32α to PR3 was determined by surface plasmon resonance. The dissociation constants were 2.65 ± 0.4 nM for urinary PR3 and 1.2 ± 0.05 nM for neutrophil-derived PR3. However, irreversible inactivation of PR3 enzymatic activity with phenylmethyl sulphonyl fluoride (PMSF) did not significantly change binding to the cytokine.(28) PR3 is a specific IL-32 binding protein, independent of its enzymatic activity. The PR3 used for surface plasmon resonance and bioassay are commercial and urine PR3, respectively. Both are in the mature form of PR3, which have full enzymatic activity. The limited cleavage of IL-32α by PR3 enhances activities of the cytokine.(28) We examined whether the MAbs neutralize the enzymatic activity of PR3 on IL-32α. An MAb that neutralizes the enzymatic activity of PR3 could be useful for blocking the inflammatory property of PR3 since the cleavage of IL-32α and other inflammatory cytokines such as IL-1β, TNFα, and IL-8 by PR3 enhanced their biological activities. However, none of the six MAbs possessed neutralizing activity (data not shown).
Next, we performed flow cytometry with the anti-PR3 MAbs (Fig. 6). The MAb clone 38 obtained the most prominent result in both THP-1 and U937 cell lines. Interestingly, although the detection signal of MAb clone 19 was much weaker than that of MAb clone 38, the former MAb stained only THP-1 cells (Fig. 6). The Western blot and flow cytometry of MAb clone 19 suggest that the membrane-bound form of PR3 from THP-1 cells is different from that from the U937 cells.
In Figure 7, the high molecular weight of PR3 was detected in both human plasma and urine although there was a little difference in molecular weight in Western blots. These high molecular weight bands may be Michaelis' complex. Serine protease inhibitors (serpins) regulate the activities of circulating proteases by acylating the serine hydroxyl at their active sites. Conformational changes are triggered in the serpin while it remains covalently linked to the protease, forming what is called Michaelis' complex, prior to deacylation and complete proteolysis of the serpin.(29,30) The high molecular weight of PR3 band in both plasma and urine may represent a Michaelis' complex formation of a covalent linkage because the reduced condition did not break down the high molecular band. In order to find out which serpin forms a Michaelis' complex with PR3, we need further study by immunoprecipiation of the high molecular bands and mass spectrometer analysis.
In this study, we have expressed recombinant huPR3 protein in E. coli and developed six MAbs anti-human PR3. The purified recombinant human PR3 (rhuPR3) protein was used to generate a panel of anti-rhuPR3 MAbs. Direct ELISA and immunoblot assays showed that five new MAbs (clones 4, 27, 32, 36, and 38) recognized both recombinant huPR3 protein and human neutrophil PR3 (commercial PR3) under reducing or non-reducing conditions. The detection limit is about 5 ng at least. Moreover, we demonstrated that anti-PR3 MAb clone 32 strongly reacted with endogenously synthesized PR3 proteins in human THP-1 and U937 cells with immunofluorescent staining. The newly developed anti-huPR3 MAbs were able to detect the endogenous PR3 in the plasma and urine by Western blot analysis.
All these data demonstrated that, in contrast to the commercial anti-hPR3 MAbs, the newly developed four MAbs have apparent advantages for the measurement of PR3 in biological samples such as plasma and tissue, and the native form of PR3 in cells. In conclusion, we have generated four anti-PR3 MAbs that are highly sensitive for detection of the endogenous PR3 protein. These MAbs should be helpful for studying the biology of PR3 and for developing diagnostic and therapeutic methods of PR3 associated inflammatory and autoimmune diseases.
Footnotes
Acknowledgments
This work was supported by a Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean government (MOST, no. R01-2006-000-10837). S.Y. was supported by the Second-Phase of BK (Brain Korea) 21 project in 2009.