
Abstract
In this report we describe the generation of a mouse monoclonal antibody (MAb) against the influenza A virus PB1-F2 protein that is derived from a +1 reading frame of the polymerase basic protein (PB1) gene segment. We further present data that the hybridoma subclone F2-6G10 produces antibodies that specifically recognize the PB1-F2 protein of H1N1 influenza virus types only. The antibody can be used for immunodetection of the PB1-F2 protein in ELISA, Western blot, immunoprecipitation, and immunofluorescence assays.
Introduction
To our knowledge no reliable antibodies against PB1-F2 are commercially available. We decided to establish mouse monoclonal antibodies directed against the PB1-F2 protein of the H1N1 IAV strain. Here we describe the properties of this monoclonal antibody.
Materials and Methods
Plasmids and purification of recombinant proteins
For expression of the GST-tag PB1-F2 protein in bacteria, the DNA fragment coding for PB1-F2 of IAV strain A/Puerto-Rico/8/34 was amplified by PCR using the pHW2000-PB1 plasmid (a kind gift from E. Hoffmann and R.G. Webster, Department of Virology and Molecular Biology, St. Jude Children's Research Hospital, Memphis, TN) as template. The obtained PCR fragment was inserted into the pGEX 6P-1 vector via SmaI/XhoI restriction sites. For expression in mammalian cells, a PB1-F2 PCR fragment was cloned in-frame into the myc-tag containing vector pCS2+MT. The integrity of the constructs was verified by sequencing before use.
The GST-PB1-F2 fusion protein or GST alone was expressed in Escherichia coli BL21. Protein expression was induced by 0.5 mM IPTG for 3 h at 37°C. Bacterial pellets were resuspended in PBS (pH 7.3), disrupted by sonification on ice, and centrifuged at 15,000 g for 30 min; supernatants were filtered through a 0.45 μm filter. Recombinant proteins were purified from these bacterial extracts by FPLC using a 1 mL GSTrap FF column (GE Healthcare, Freiburg, Germany) according to the supplier's instruction. PBS (pH 7.3) was used as binding buffer, and 50 mM Tris-HCl (pH 8.0), with 15 mM reduced glutathione, was used as elution buffer. Obtained protein fractions were analyzed by SDS-PAGE followed by Coomassie staining. Fractions containing recombinant proteins were then combined, dialysed against PBS (pH 7.3), and analyzed again by SDS-PAGE and Coomassie staining before use.
Immunization of mice and establishment of hybridoma
Eight-week-old female BALB/c mice were immunized intraperitoneally (i.p.) with 50 μg of GST-PB1-F2 chimeric protein. Four weeks after priming, mice were boosted five times in 2-week intervals by i.p. injection of 50 μg GST-PB1-F2 protein per mouse. Spleen cells of immunized mice were washed two times with PBS, mixed with logarithmically growing SP2/0 myeloma cells in a relation of 5:1, and fused using polyethylene glycol 1500 (783641, Roche, Mannheim, Germany). Fused cells were resuspended in RPMI1640 medium supplemented with 20% FCS, 10 U/mL IL-6 (1299972, Roche), OPI media supplement (O5003, Sigma-Aldrich, Taufkirchen, Germany), and HAT media supplement (H0262, Sigma-Aldrich) and plated into 96-well tissue culture plates with 1.2 × 105 cells per well in a volume of 200 μL. After cultivation at 37°C with 7.5% CO2 for 10 days, supernatants of mature clones were analyzed by ELISA for presence of anti-PB1-F2 and anti-GST antibodies.
Enzyme-linked immunosorbent assay
ELISA plates were coated o/n at 4°C with 50 μL of purified GST or GST-PB1-F2 proteins (5 μg/mL), blocked for 1 h at room temperature with 1% BSA, and incubated for 1 h at room temperature with 50 μL of hybridoma supernatants. After washing with PBS, ELISA plates were further incubated for 30 min with goat anti-mouse antibodies labeled with horse radish peroxidase (HRP), washed three times with PBS, and incubated with the ABTS substrate (1112597, Roche, Mannheim, Germany) for an additional 30 min and finally measured photometrically at 405 nm.
Ig isotyping
The isotyping of obtained mouse monoclonal antibodies was performed using the Roche Antibody Isotyping Kit (1293027, Roche).
Cell lines, cDNA transfection, and viral infection
Madin-Darby canine kidney (MDCK) cells were grown in MEM (minimal essential medium), HUVEC (human umbilical vein endothelial cells) in a mix of endothelial growth medium (EGM) and M199 with Earls salts, HEK 293 cells in DMEM (Dulbecco's MEM), and SP2/0 myeloma cells in RPMI1640 medium. All culture media were supplemented with 10% heat-inactivated fetal bovine serum (FBS).
NIH 3T3 fibroblasts were transfected with PEI (polyethylenimine) and other cells used in this work with Lipofectamine 2000 (Invitrogen) as previously described.(10) For IAV infection cells were washed with PBS and incubated with the indicated multiplicity of infection (moi) diluted in PBS/BSA (PBS containing 0.2% BSA, 1 mM MgCl2, 0.9% CaCl2, 100 U/mL penicillin, and 0.1 mg/mL streptomycin) for 30 min at 37°C. After this incubation, inoculums were aspirated and cells were washed with PBS and further incubated for 6–24 h in either MEM or D-MEM containing 0.2% BSA and antibiotics.
Immunoprecipitation and immunoblotting
At time points indicated, cells were lysed with Triton X-100-containing buffer (20 mM Tris-HCl (pH 7.4), 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 50 mM sodium glycerophosphate, 5 μg mL−1 aprotinin, 5 μg mL−1 leupeptin, 1 mM sodium vanadate, and 5 mM benzamidine) for 20 min. Cell lysates were centrifuged at 10,000 g for 10 min, supernatants collected, and protein amounts determined using BCA Protein Assay Kit (Thermo Scientific, Bonn, Germany). Twenty μg of total cell lysates were separated by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted on nitrocellulose membranes (NCM) for Western blot analysis.
For immunoprecipitation, the supernatant of the hybridoma subclone F2-6G10 was incubated for 1 h at room temperature with either protein A or G agarose beads to immobilize the antibodies. After washing once with lysis buffer, the agarose beads were incubated with 500 μL of cell lysates containing 300 μg of total proteins for 2 h at 4°C and washed three times with lysis buffer. Immunoprecipitated proteins were separated by SDS-PAGE, blotted onto NCM, and analyzed further by Western blotting. Therefore NCMs were blocked with a 5% solution of skim milk in TBST buffer containing 0.2% Triton X-100 for 1 h at room temperature, incubated for 1 h with primary antibodies, and washed. After incubation with HRP-labeled secondary antibodies, the proteins were visualized by standard ECL chemiluminescence and analyzed with a Lumi-Imager using the AIDA software. In addition to the monoclonal antibodies obtained and analyzed here, the following antibodies were used: (1) MAb against the NS1 protein of influenza A viruses (clone NS1-23-1, produced in the Institute of Molecular Virology, Muenster, Germany). The antibody was used to detect the efficiency of infection, as it recognizes all IAV subtypes tested so far; (2) Anti-GST MAb (clone G3E7, produced in the Institute of Molecular Virology, Muenster, Germany); (3) anti-myc MAb (clone 9E10). As secondary antibodies, sheep anti-mouse IgG-HRP conjugates (1:2500; Dianova, Hamburg, Germany) were used.
Immunofluorescence studies
Mouse NIH 3T3 fibroblasts were transiently transfected with a myc-tagged PB1-F2 plasmid. At 24 h post-transfection, cells were trypsinized, plated on fibronectin-coated glass coverslips for 2 h, fixed with 2 % PFA, permeabilized with 0.2% Triton X-100, and stained for expression of PB1-F2, either with the specific monoclonal antibody or with the anti-myc MAb (clone 9E10) or rabbit polyclonal anti-myc serum. The F-actin was visualized with Alexa-488 labeled phalloidin. As secondary antibodies, either Cy3-labeled goat anti-mouse or Alexa-488 conjugated goat anti-rabbit antibodies were used. Cell images were taken using an Axiovert 2000 ApoTome microscope with an AxioCam digital camera and AxioVision software (Zeiss, Jena, Germany).
Results and Discussion
Establishing anti-PB1-F2 hybridoma subclones
PB1-F2 is a small protein consisting of only 87 amino acids. Thus we expressed the polypeptide in bacteria as a GST-fusion protein, which enables its purification by passing bacterial lysate over a glutathione-sepharose bead column. The GST-tag was not cleaved off but the complete GST-PB1-F2 fusion protein was used as an antigen. Twenty-four clones that were grown after fusion of splenocytes with myeloma SP2/0 and were revealed by ELISA as positive for GST-PB1-F2, but not for GST alone, were transferred into 6-well plates. After reaching confluency, the supernatants of the clones were tested for anti-PB1-F2 and anti-GST activity by Western blot analysis. HEK 293 lysates containing recombinantly expressed myc-tagged PB1-F2 and the bacterially expressed GST protein were resolved by SDS-PAGE and served as targets for hybridoma supernatants. Eight clones, the supernatants of which recognized the myc-tagged PB1-F2 but not the GST in immunoblotting, were subcloned by limiting dilution; the subclones were further tested for PB1-F2 specificity by ELISA and Western blot assays. Subclones that have produced antibodies working in both ELISA and Western blot assays with no cross-reactivity to GST protein alone were propagated and used further as established hybridoma cell lines.
Binding specificity in Western blot analysis
We then wondered whether the obtained monoclonal antibodies would also recognize PB1-F2 proteins of other influenza virus strains. Therefore MDCK or HUVEC cells were infected with different IAV strains: A/Puerto-Rico/8/34 (H1N1), A/Victoria/3/75 (H3N2), A/FPV/Bratislava/79 (H7N7), A/Thailand/KAN-1/2004 (H5N1), or left uninfected. Sixteen h post-infection, total cell lysates were studied for expression of the different PB1-F2 proteins using supernatants of the hybridoma subclones as primary antibodies in immunoblotting. As a positive control, a recombinant myc-tagged PB1-F2 protein (H1N1) was used that was transiently expressed in HEK 293 cells after their transfection with pCS2+MT/PB1-F2 plasmid. Western blot analysis revealed that all eight tested subclones recognized the PB1-F2 protein from H1N1 influenza virus only but not from other virus strains. Figure 1A shows the typical specific detection of the 14 kDa PB1-F2 protein derived from PR8 (H1N1) virus strain. In this case the subclone F2-6G10 was used. To ensure that the cells were indeed infected and IAV proteins were expressed, the same cell lysates were analyzed by another MAb directed to the 26 kDa NS1 IAV protein. The anti-NS1 antibody was also produced in our lab against the NS1 protein of the PR8 strain (H1N1), but these MAbs (clone NS1-23-1) specifically recognized the NS1 of all IAV strains so far analyzed. Western blot analysis with this anti-NS1 MAb confirmed that both MDCK and HUVEC cells were efficiently infected with the different IAV subtypes used and that the obtained anti-PB1-F2 MAbs indeed recognize only the PB1-F2 protein of the H1N1 origin. Therefore in our further experiments we characterized MAbs of only one subclone, the F2-6G10. Ig isotyping of this monoclonal antibody identified it as an antibody of the IgG2b subclass with the kappa light chain.
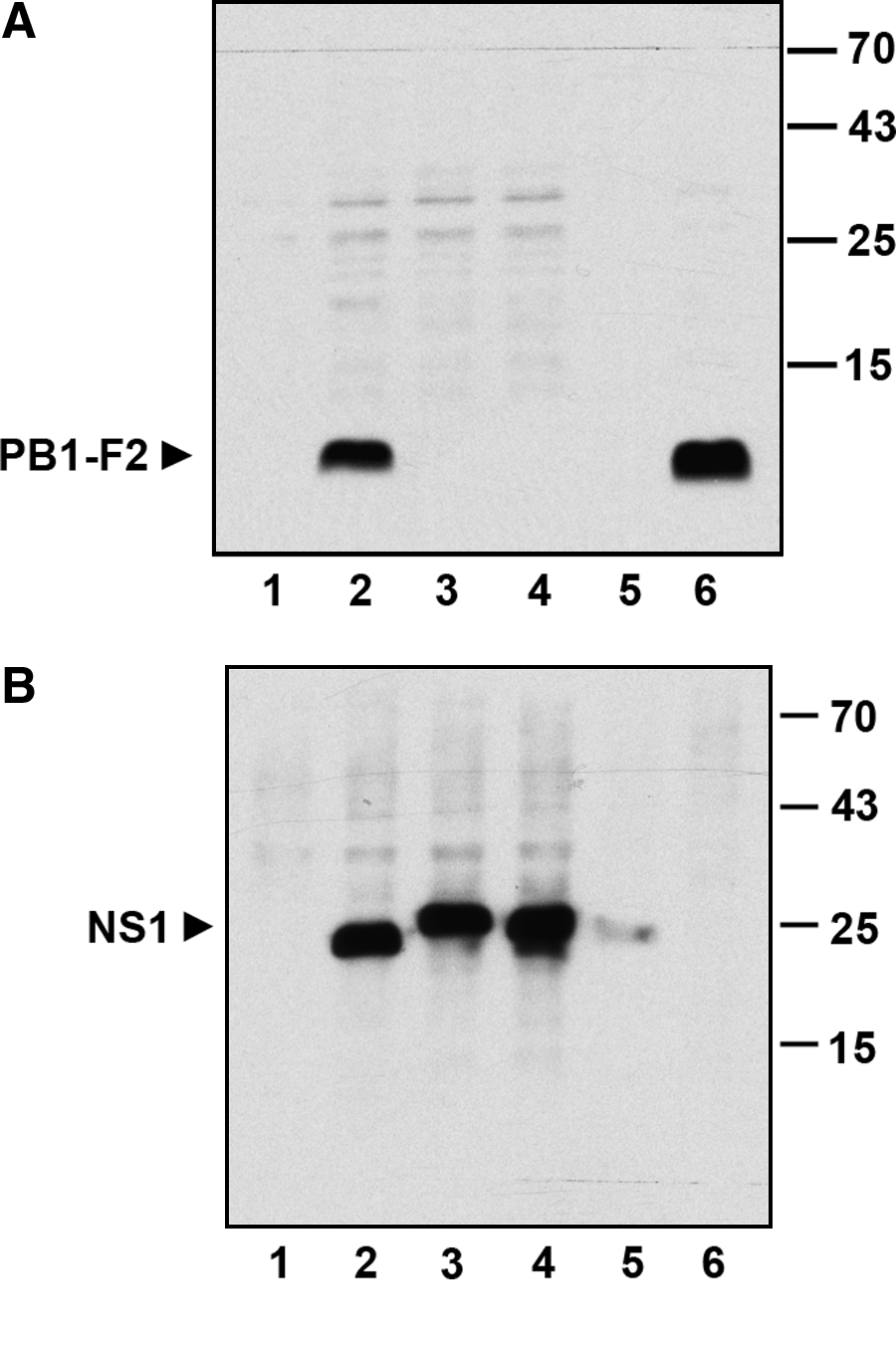
Western blot analysis of PB1-F2 by F2-6G10 MAb. MDCK or HUVEC cells were left uninfected (lane 1) or were infected with different IAV strains: lane 2, A/Puerto-Rico/8/34 (H1N1); lane 3, A/Victoria/3/75 (H3N2); lane 4, A/FPV/Bratislava/79 (H7N7); lane 5, A/Thailand/KAN-1/2004 (H5N1). Cell lysates were separated by SDS-PAGE and after electroblotting onto nitrocellulose the expression of viral proteins was analyzed by Western blotting with F2-6G10 MAb (
Immunoprecipitation studies
To test whether F2-6G10 monoclonal antibodies can also precipitate the PB1-F2 protein, the hybridoma supernatant was incubated with protein A or protein G agarose beads for 1 h at room temperature. As can be seen from Figure 2, the antibody binds to both proteins A and G, but with a higher efficiency to protein G. According to this, more PB1-F2 protein was immunoprecipitated by the antibody when it was coupled to protein G agarose beads. The precipitation of PB1-F2 by the antibody was specific, as the PB1-F2 protein did not bind to protein A or G agarose beads that had not been precoated with hybridoma supernatant. Thus, the monoclonal antibody F2-6G10 recognizes not only the SDS-denatured viral PB1-F2 protein but also the intact non-denatured one.
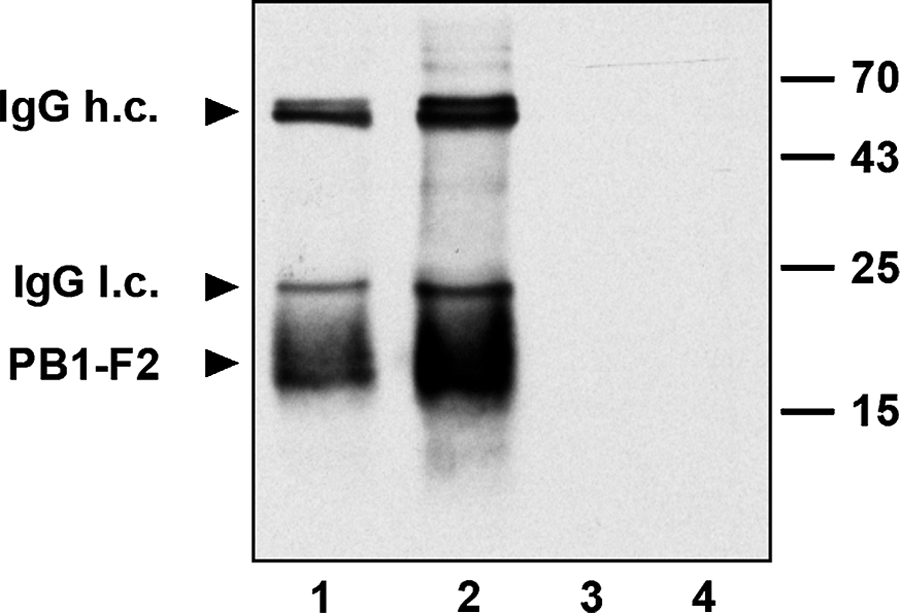
Immunoprecipitation of PB1-F2 protein by F2-6G10 MAb. The myc-tagged PB1-F2 protein (H1N1) was transiently expressed in HEK 293 cells and immunoprecipitated with the MAb F2-6G10 coupled either to protein A (lane 1) or protein G agarose beads (lane 2). Immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted using anti-myc MAb (9E10) for PB1-F2 detection. Lysates from transfected samples that were coupled to protein A (lane 3) or protein G agarose beads (lane 4) alone served as specificity controls.
Immunofluorescence studies
Finally we analyzed whether the F2-6G10 MAb can also be used for immunodetection of the PB1-F2 protein in fixed mammalian cells and whether it will specifically detect the PB1-F2 only. To this end NIH 3T3 fibroblasts were transfected with a plasmid coding for myc-tagged PB1-F2 of PR8 origin (H1N1). At 24 h post-transfection, the cells were trypsinized and allowed to spread for 2 h on fibronectin precoated glass coverslips. After the cells were fixed with paraformaldehyde and permeabilized, they were incubated for 1 h at room temperature with F2-6G10 hybridoma supernatant and the PB1-F2 protein was finally detected by a Cy3-labeled goat-anti-mouse antibody. To visualize the shape of all cells plated, they were counterstained with Alexa-488 labeled phalloidin and with DAPI. Figure 3 clearly shows that the F2-6G10 MAb does not stain all but only transfected cells and that the PB1-F2 protein is localized exclusively in the cytosol with a prevalent perinuclear stain. Staining of transfected NIH 3T3 cells with the anti-myc MAb 9E10 revealed a very similar staining to that of the F2-6G10. To further underline the specificity of our monoclonal antibody, the transfected NIH 3T3 cells were co-stained with the anti-PB1-F2 MAb F2-G610 antibody and a rabbit polyclonal anti-myc serum. Both antibodies were able to stain only transfected cells and recognized absolutely the same structures within the transfected cells. To ensure that the F2-6G10 MAb also recognizes the native PB1-F2 protein after infection of cells with influenza viruses, A549 human lung epithelial cells were infected with PR8 IAV (H1N1), fixed 5 or 10 h post-infection, and stained for PB1-F2 expression with our monoclonal F2-6G10 antibody. Figure 4 demonstrates that the antibody also recognizes the virally expressed PB1-F2 protein. The results further show that the antibody staining is highly specific and that the viral PB1-F2 protein is localized mainly in the cytosol, being concentrated in structures resembling mitochondria, which corresponds to already published data. Thus, our data unambiguously show that the F2-6G10 MAb is suitable for immunofluorescence studies of the influenza viral PB1-F2 protein and that it does not show a significant background staining.
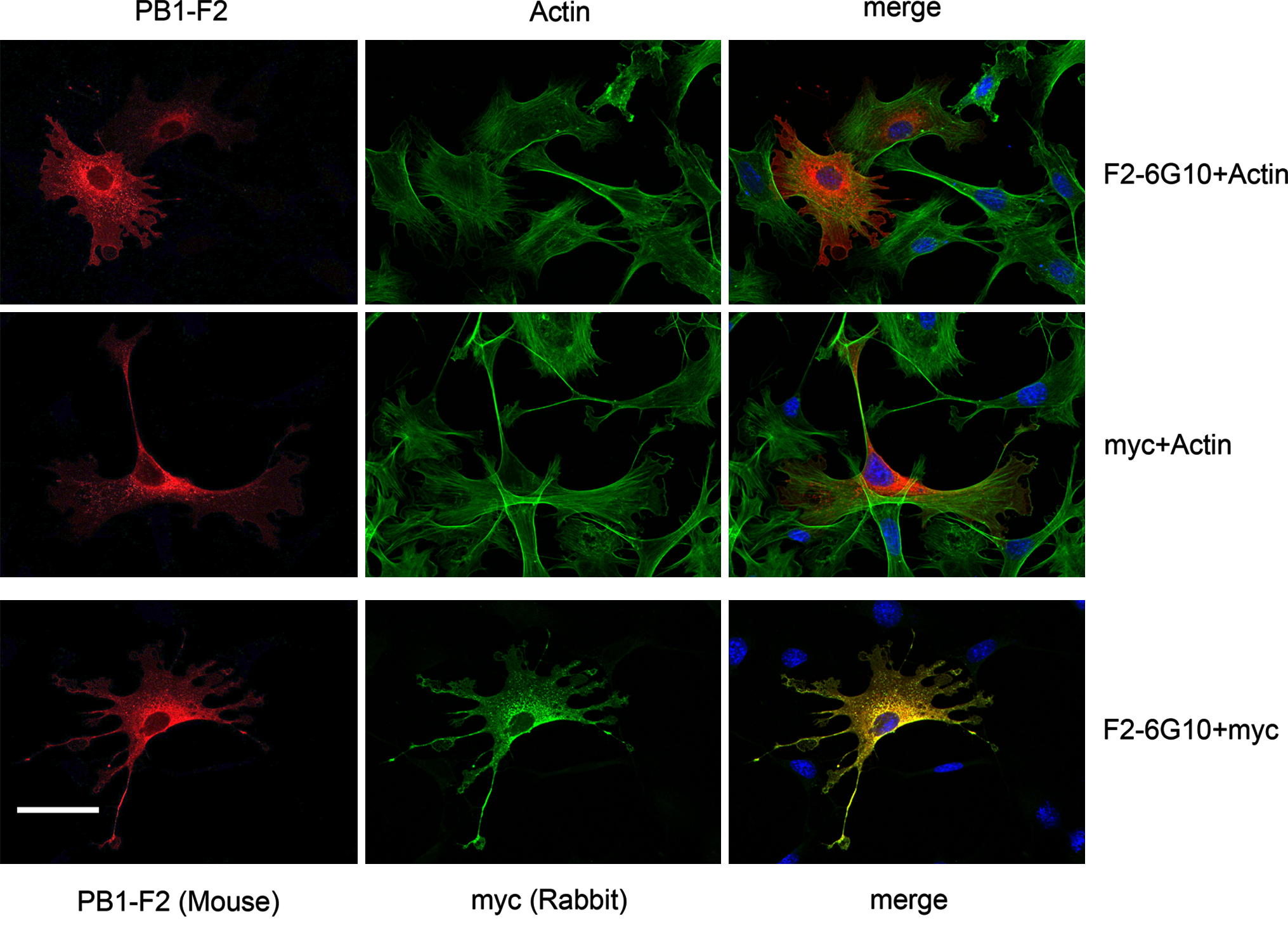
Detection of recombinant PB1-F2 protein with F2-6G10 MAb by immunofluorescence. myc-tagged PB1-F2 (H1N1) protein was transiently expressed in mouse NIH 3T3 fibroblasts and its expression was analyzed with the PB1-F2 specific (F2-6G10) or myc-tag-specific (9E10) MAbs (upper and middle panels, respectively), or with the MAb F2-6G10 and rabbit anti-myc serum (lower panels) to show that the anti-PB1-F2 MAb indeed specifically recognizes the recombinant PB1-F2 protein only. To visualize all cells, the nuclei and actin cytoskeletons were revealed by DAPI and Alexa-488-labeled phalloidin, respectively.
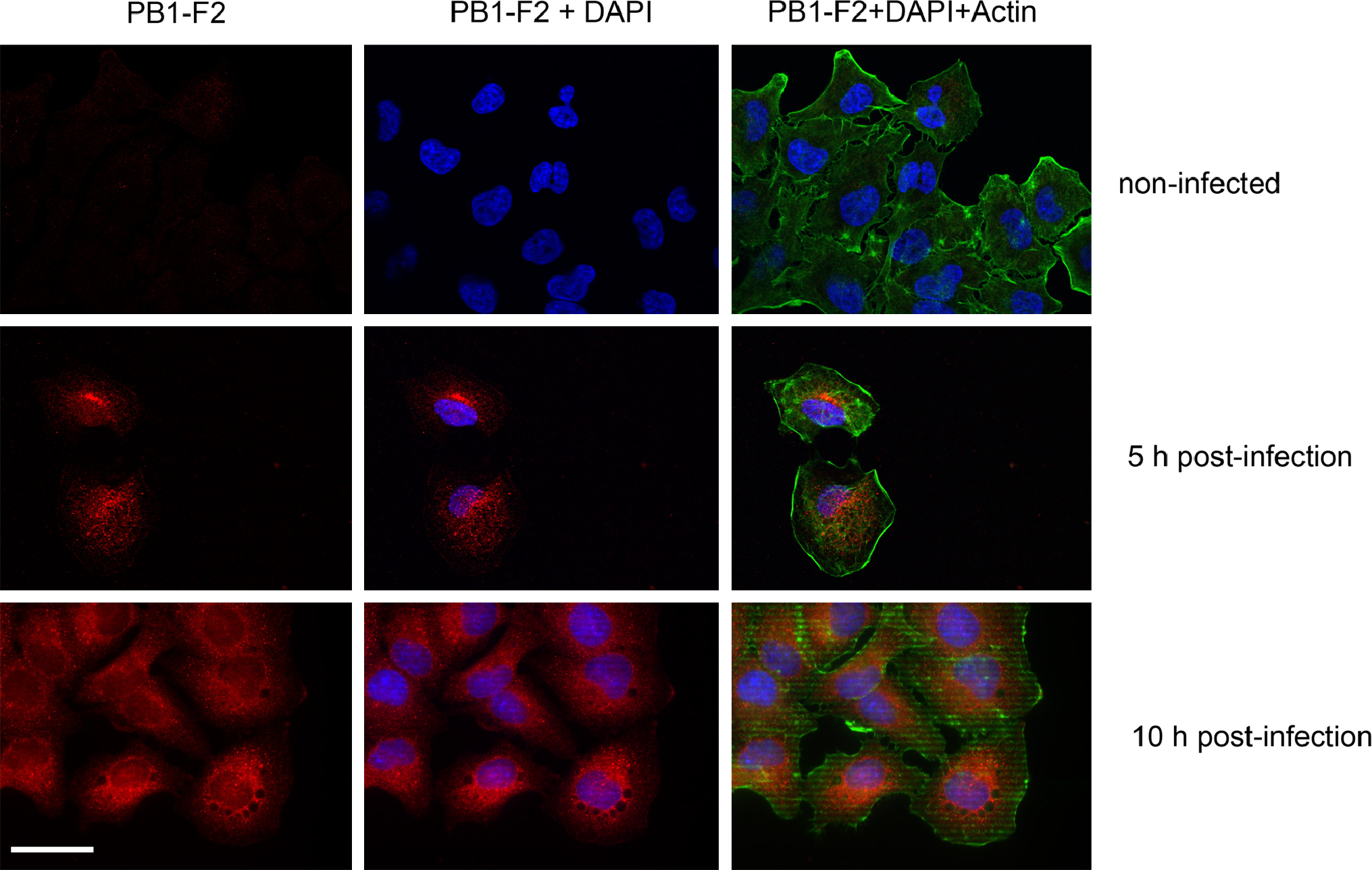
The MAb F2-6G10 recognizes the native PB1-F2 protein in viral infected cells. Human A549 lung epithelial cells were infected with A/Puerto-Rico/8/34 (H1N1) influenza virus for different times and analyzed for expression of viral PB1-F2 protein. Cells were co-stained with DAPI and Alexa-488-labeled phalloidin to visualize their shape.
In summary, we produced a mouse monoclonal antibody that specifically recognizes the H1N1 influenza virus protein PB1-F2 and can be used in ELISA, immunoblotting, and immunoprecipitation, as well as in immunofluorescence techniques.