
Abstract
Efficient clearance of cells undergoing apoptotic death is crucial for normal tissue homeostasis and for the prevention of autoimmunity. Engulfment of apoptotic cells is finely regulated by a highly redundant system of receptors and bridging molecules. We developed a rapid and efficient method for the identification of human natural IgM antibodies and isolated some natural human monoclonal IgM antibodies specific to the apoptotic cells. Among them, AC34 IgM bound to early apoptotic cells and promoted phagocytosis of apoptotic Jurkat cells by human monocyte-derived macrophage (HMDM). AC34 IgM recognized phosphatidylserine (PS), which means PS might be a possible molecule recognized by AC34 IgM on the surface of apoptotic cells and AC34 IgM might be a possible candidate of bridging molecules between the PS and phagocytes. The sequences of VH and VL of AC34 were almost the same with their germline counterpart. Our experiments suggest a role of natural IgM as an opsonin in the clearance of early apoptotic cells.
Introduction
Among various methods for making human monoclonal antibodies, Epstein-Barr virus (EBV) has been used to transform B lymphocytes and derive a population of cells secreting specific antibodies of interest.(1) Isolating stable populations of these monoclonal cells, however, has proven very difficult. In the 1990s, introduction of electrofusion under strong hypo-osmolar condition allowed the efficiency of hybridoma formation to greatly increase. Perkins and Foung combined both techniques and developed a new method to immortalize specific antibody-producing cells by fusing secreting EBV-activated lymphocytes to mouse-human heteromyeloma cell lines with electrofusion under hypo-osmolar condition.(2) In this study, the method by Perkins and Foung was adopted to acquire human monoclonal hybridoma clones, which secrete autologous natural IgM antibodies.
Natural antibodies have been acknowledged and thought to play an important role in the immune system for decades. Natural antibodies are mainly IgM isotype and poly-reactive to a variety of self and non-self antigens. They are secreted by B1 and splenic marginal zone B cells in mice. Recent studies apparently revealed that these antibodies are involved in many diverse processes, including primary immune defense against infective organelles, prevention of autoimmunity, and surveillance against cancers; it was further revealed that they affect B-cell development and help to maintain T-cell memory.(3) The mechanism of autoimmunity caused by IgM deficiency has not been clearly defined but it might be related to the clearance of apoptotic debris/cells. Apoptotic cells provide a potential source of immunogens for the development of autoimmunity. Boes and colleagues bred Sμ mice (i.e., serum IgM-deficient mice) with the lupus-prone MRL-lpl strain. The lpl mice showed high SLE features that were significantly higher in the lpl/Sμ mice.(4) Elkon and colleagues demonstrated that natural IgM recognize dying cells through the Fab’ portion of the antibody and IgM bind to the lysophopholipid form of phosphatidylcholine.(5) Moreover, the action of C1q, which is a key component of opsonization of apoptotic cells, is entirely dependent on the binding of IgM to the apoptotic debris.(6) All of this evidence suggests that IgM plays an important role in the clearance of apoptotic cells directly or through complement activation.
Specific recognition and removal of apoptotic cells by phagocytes are very complicated processes and not completely understood. Many receptors, adaptors, and chemotactic molecules are reportedly involved in these processes.(7) Recently, mice monoclonal IgM antibody (3B4), which recognizes apoptotic cells and promotes phagocytosis, was identified and characterized.(8) 3B4 IgM binds to late apoptotic cells and promotes phagocytosis of apoptotic cells in a complement-independent manner. This suggests that IgM antibodies can be a bridging molecule between apoptotic cells and macrophages. Although most of the previous studies have suggested that natural IgM antibodies promote the uptake of apoptotic cells, the molecular structure and characteristics of those natural IgM antibodies in humans have not been clearly defined. In this study, we developed a rapid and efficient method for the identification of human natural IgM antibodies and isolated some natural IgM-producing hybridoma clones. Among them, AC34 bound to early apoptotic cells and promoted phagocytosis of apoptotic Jurkat cells by human monocyte-derived macrophage (HMDM). The sequences of VH and VL of AC34 were almost the same as their germline counterpart.
Materials and Methods
Cell culture
H73C11, a mouse-human heteromyeloma cell line, was cultured in the RPMI1640 (Gibco, Grand Island, NY) supplemented with 10% FBS (Gibco), 2 mM L-glutamine (Gibco), 2 mM sodium pyruvate (Sigma, St. Louis, MO), 1% non-essential amino acid (Gibco), 50 μM 2-Mercaptoethanol (Gibco), and 1% penicillin/streptomycin (Gibco). Jurkat cells and B95-8, an EBV-secreting marmoset cell line, were cultured in RPMI1640 supplemented with 10% FBS. HepG2 cells were cultured in low glucose DMEM media supplemented with 10% FBS. Jurkat, B95-8, and HepG2 cells were purchased from ATCC (Manassas, VA).
Generation of human hybridoma
Lymphocytes were isolated from abdominal lymph nodes dissected from five different hepatoma patients during lobectomy. T cells were depleted by AET (Sigma)-treated sheep RBCs, and isolated B cells were activated with IMDM media (Gibco) containing 5% B95-8 supernatants (vol/vol) and 30% FBS in the 96-well round-bottom culture plate (Falcon, Oxnard, CA) for about 20 days. An immunofluorescence assay with unfixed, live HepG2 cells detected positive wells containing apoptotic specific IgM antibodies. The electrofusions of specific EBV-activated cells with H73C11 using Multiporator (Eppendorf, Hamburg, Germany) were done, and produced human hybridoma cells underwent HAT selection (Sigma) for about 20 days. IgM-producing hybridomas were screened by ELISA. An immunofluorescence assay with unfixed, live HepG2 cells detected positive hybridomas, which secrete apoptosis-specific IgM antibody, and they were cloned by limiting dilution.
Live cell immunofluorescence assay
HepG2 cells were seeded to the 96-well flat-bottom culture plate (Falcon) at the count of 3 × 105 cells/well. Supernatants of EBV-activated B cells or hybridomas were 4-fold diluted with PBS/5% dextrose solution mixture (2:1 vol/vol) containing 1% BSA. Fifty μL of diluted supernatants were incubated with HepG2 cells for 30 min at 4°C. Each well was washed by 200 μL cold PBS/5% dextrose solution mixture three times with multi-channel pipette (Eppendorf ). Fifty μL of FITC conjugated goat anti-human IgM Fcμ (Jackson Immunoresearch, West Grove, PA), which were 1:100 diluted by PBS/5% dextrose solution mixture (2:1 vol/vol) containing 1% BSA, were added to each well and incubated for 30 min at 4°C. Each well was washed again with 200 μL cold PBS/5% dextrose solution mixture three times with multi-channel pipette. After final washing, 50 μL of 4% paraformaldehyde (Sigma) was added to each well and incubated for 10 min at room temperature. Cells were observed with inverted fluorescent microscopy.
Apoptosis induction and binding analysis of AC32, AC34, AC39 IgM to apoptotic cells
Apoptosis was induced to the Jurkat cells by the addition of 1 μM staurosporine, and cells were incubated at 37°C in 5% CO2 for different time periods (0, 4, 10 h). The percentages of early and late apoptotic cells were quantified by flow cytometry analysis using Annexin V and propidium iodide (PI) according to the manufacturer's instructions (Becton Dickinson, San Jose, CA). Apoptotic cells at different time points were challenged with AC32, AC34, AC39, and control IgM (#31146, Pierce, Rockford, IL) separately for 30 min at 4°C. After washing with cold PBS three times, cell-bound IgM antibodies were analyzed by flow cytometry after addition of FITC conjugated goat anti-human IgM Fcμ antibody (Jackson Immunoresearch).
Reactivity of AC34 IgM to phospholipids
AC34 IgM against phosphatidylserine (Avanti, Alabaster, AL) and phophatidylcholine (Avanti) antigens was analyzed by ELISA. Phopholipid antigens were dissolved in ethanol, and 100 μL of diluted phospholipids (0.3 μg/mL) were added to each well of ELISA plates with hydrophobic surface (Thermo Lab Systems, Waltham, MA), then allowed to dry overnight at 4°C. The wells were washed three times with PBS, and blocked with PBS containing 3% BSA for 2 h at room temperature. Fifty μL of diluted antibodies (10 μg/mL) was then added and incubated for 2 h at room temperature. After being washed three times, the wells were incubated with 50 μL of HRP-conjugated goat anti-human IgM (diluted 1:10,000, Jackson Immunoresearch). After being washed three times again, 50 μL of substrate TMB (Pierce) were added to develop color. Control IgM (Pierce #31146) was used as negative control. OD was measured at 450 nm with an ELISA reader (Titertek Multiskan MCC/340, ICN Phamaceutical, Zoetermeer, The Netherlands).
Isolation and culture of human monocyte-derived macrophages
Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy blood donors by density-gradient centrifugation on Ficoll-Hypaque (Sigma). Monocytes were purified by adherence to plastic in RPMI1640 (Gibco) supplemented with 10% FBS (Gibco). 2.5 × 106 PBMC cells were seeded into each well of 24-well culture plates (Falcon) and, after one day, non-adherent cells were removed by several washes with PBS. Freshly isolated monocytes were allowed to differentiate into HMDMs in complete RPMI 1640 supplemented with 10% FBS and human recombinant macrophage colony stimulating factor (100 ng/mL, Sigma) for 6 days before phagocytosis assay.(10)
Phagocytosis assay
Well-growing Jurkat cells were labeled with red fluorescent aliphatic dye PKH26 (Sigma) before induction of apoptosis. After incubation with 1 μM staurosporine for 4 h, apoptotic cells were co-cultured with HMDMs at 2:1 ratio in the presence of 20 μg/mL of AC34, 20 μg/mL of control IgM, and media only for 30 min. The wells were then washed two times with PBS to eliminate most of non-phagocytosed apoptotic cells. To quench fluorescence of remaining non-phagocytosed cells, 0.2% Trypan blue (Sigma) was added to each well and incubated for 10 min. The wells were then washed two times again with PBS and loaded on the ice to stop phagocytosis. The cells were observed by inverted fluorescent microscopy and three different areas per well were photographed for cell counting. The percentage of HMDM cells that contain red apoptotic cells among total HMDM cells was calculated.
Sequencing of AC34 VH and VK cDNA
Total cellular RNA was extracted from AC34 hybridoma cells using TRIzol reagent (Gibco-BRL, Life Technologies, Gaithersburg, MD). Reverse transcription reaction was performed using a kit from Promega (San Luis Obispo, CA), with total RNA as template and oligo-dT as a primer. The resulting cDNA was used to amplify VH and VK with Novagen human Ig primer set (Novagen, San Diego, CA) following the manufacturer's manual. The variable region genes of AC34 IgM were analyzed by IMGT/V-QUEST tool (
Results
Isolation of hybridoma clones that secrete IgM antibodies specific to the apoptotic cells
Using immunofluorescence assay with live HepG2 cells, eight hybridoma clones that secrete autologous IgM were successfully isolated and cloned from the five series of electrofusions (AC28, AC32, AC32, AC33, AC34, AC35, AC39, and AC48). Their staining patterns in the live immunofluorescence assay were diverse, and isolated autologous IgM antibodies were classified into four groups according to their staining patterns. Group A antibodies (AC28, AC33, AC35) showed membrane staining pattern in all the cells (Fig. 1A). Group B antibodies (AC32, AC39) showed positive staining only in the small, shrunken cells (Fig. 1B). Group C antibodies (AC31, AC48) showed membrane staining pattern in some cells only (Fig. 1C). Finally, group D antibodies (AC34) showed a mixed staining pattern of groups B and C (Fig. 1D). To check if group B and D antibodies (AC32, AC34, and AC39) are staining dying cells, HepG2 cells seeded in the 96-well culture plates were dual stained with both autologous IgM antibodies and propidium iodide (PI). PI staining was co-localized with some of group B and D IgM stained cells (Fig. 1E, F). The number of HepG2 cells stained by group B and D antibodies was increased after apoptosis induction by 2 μM staurosporine for 4 h (data not shown).

Diverse autologous staining patterns of hybridoma supernatants in the live immunofluorescent assay. Eight human monoclonal IgM antibodies were successfully isolated and cloned by human hybridoma methods. Isolated human IgM antibodies were classified into four groups according to their autologous staining patterns in live immunofluorescent assay. (
AC32 and AC39 bind to late apoptotic cells and AC34 binds to both early and late apoptotic cells
Well-growing Jurkat cells were committed to apoptosis by incubation with 1 μM staurosporine in serum-free RPMI 1640 media. After 4 and 10 h treatments, different percentages of early or late apoptosis were observed (Table 1). Annexin-V+PI- cells were defined as early apoptotic phase and annexin-V+PI+ cells defined as late apoptotic phase. Without staurosporine treatment (0 h), about 15% of cells were detected in the early or late apoptotic phase and most of the cells were viable. After 4 h treatment of 1 μM staurosporine, most of cells (90.75%) were in the early apoptotic phase and the percentage of late apoptotic cells increased a little (from 5.1 to 8.85%). After 10 h treatment of 1 μM staurosporine, 80.35% of cells were in the late apoptotic phase. AC32 and AC39 IgM scarcely bound to Jurkat cells at the early apoptotic phase (4 h incubation with staurosporine) but bound strongly to the cells at the late apoptotic phase (10 h incubation with staurosporine). AC34, however, bound well to both the early and late phase apoptotic cells and the binding was increased at the late apoptotic phase. Control IgM did not bind to Jurkat cells of any apoptotic phases (Fig. 2A).
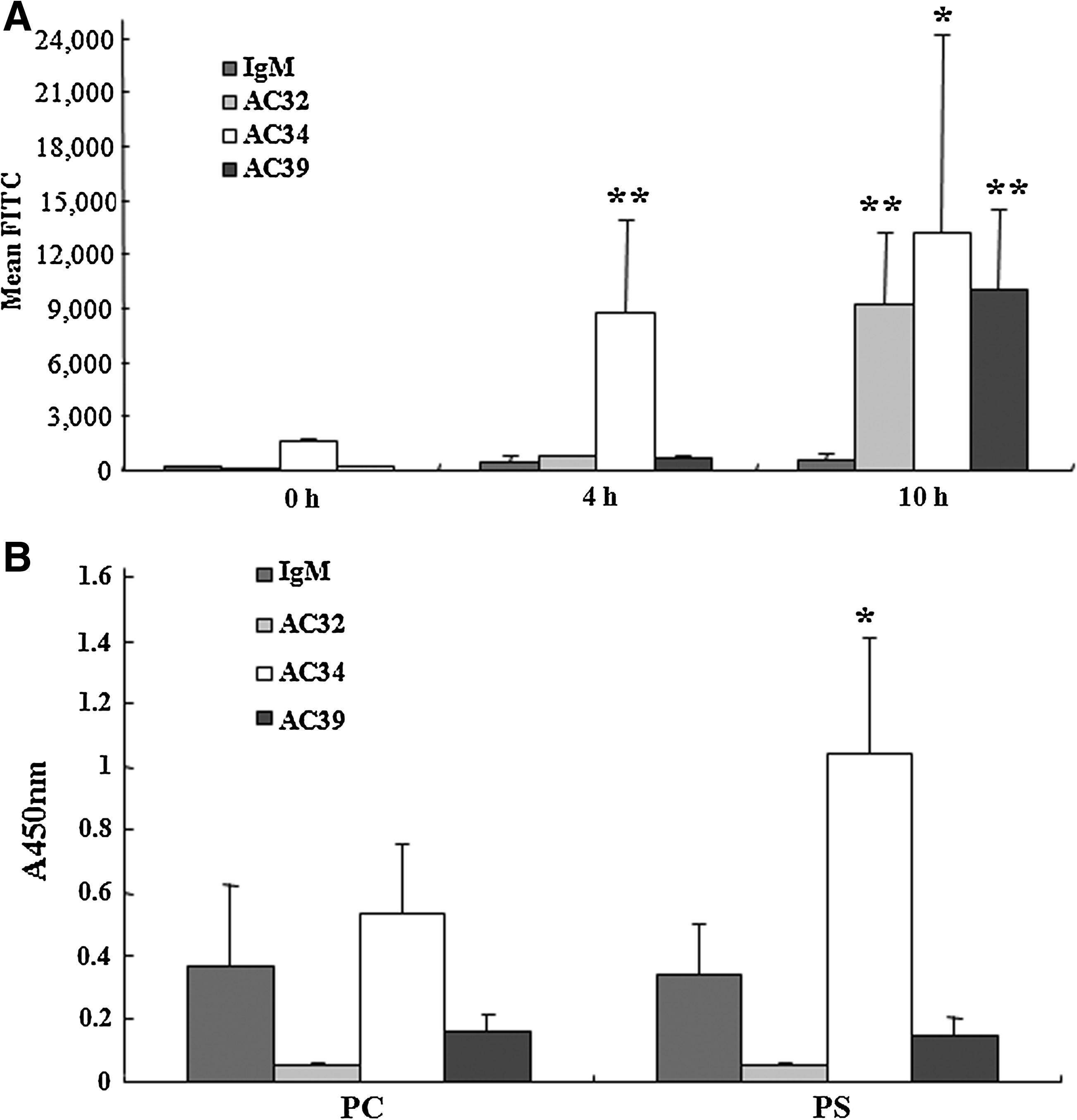
(
After incubation with 1 μM staurosporine for 0, 4, and 10 h, Jurkat cells were stained with annexin V and Pi and were analyzed by flow cytometry. Data are expressed as the means ± SD in the triplicate experiments. Apoptotic cells at the 4 h treatment were composed of mainly early phase apoptotic cells, while cells treated for 10 h were almost in the late phase.
AC34 IgM binds to phosphatidylserine antigen
AC32, AC34, and AC39 IgMs were tested for reactivity with phosphatidylcholine (PC) and phosphatidylserine (PS). AC34 IgM had definite binding to PS but AC32, AC39, and control IgMs did not show any significant binding to PC and PS (Fig. 2B). This result corresponds with the fact that AC34 IgM bound to the early apoptotic cells, which display phosphatidylserine extracellularly.
AC34 IgM increased phagocytosis of early apoptotic cells by HMDMs
Because the externalization of PS is a key event of early apoptosis, we selected AC34 IgM for the apoptotic cell phagocytosis experiment and assayed the degree of internalization of AC34-bound apoptotic cells into macrophages. Figure 3 shows the internalization of apoptotic cells increased significantly as a consequence of AC34 IgM binding. AC34 promoted the uptake of early apoptotic Jurkat cells by 15% from media only group and by 19% from control IgM group. There was no significant increase of internalization of apoptotic cells in control IgM group.
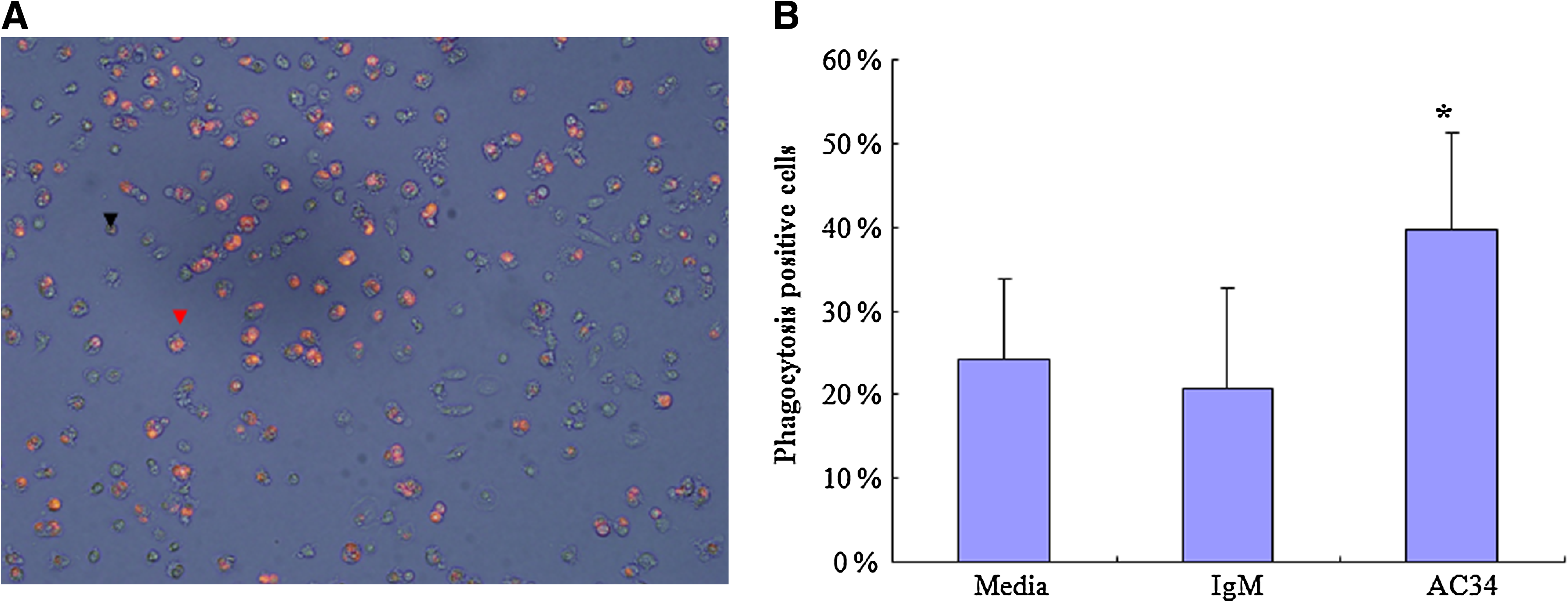
AC34 promoted the uptake of apoptotic Jurkat cells by human monocyte derived macrophages. (
AC34 IgM sequence reveals germline encoded Ig VH and VK genes
Analysis of the nucleotide sequences from the VH of AC34 hybridoma showed that AC34 VH sequences were 100% identical to that of germline IGVH4-34*01 (Fig. 4A). The AC34 VK gene was very close to its germline counterpart IGKV3-15*01 with only one nucleotide difference occurring in FR1 (Fig. 4B) that resulted in one amino acid change. It could exclude the possibility that this mutation might represent a germline gene polymorphism.

(
The nucleotide and predicted amino acid sequence from junctional regions of heavy (H) and light (L) chains are shown in Figure 4C. For the H chain of AC34 hybridoma, D region was IGHD3-22*01 and J region was IGHJ5*02. N1 addition (12 nucleotides) and N2 addition (12 nucleotides) exist at the V-D and D-J junctions. The frequency of nucleotide deletions at the V-D-J junctions was measured by the number of nucleotide deleted at the 5′-terminal end of the JH segment. No 5′-JH nucleotide loss was observed in AC34 hybridoma. For the L chain of AC34 hybridoma, J region was IGKJ1*01. At the V-J junction, N addition (two nucleotides) and P addition (one nucleotide) were observed but no nucleotide deletion was detected.
Discussion
In this study, we transformed B cells with Epstein-Barr virus at the first step to screen autologous antibodies. Because EBV-activated cells are very unstable, they need to be fused with myeloma cells to form immortalized hybridoma. EBV-activated B cells could be fused as soon as target antibodies were detected in the 96-well culture plates by immunofluorescence assay, because as few as 1 × 105 EBV-activated B cells could be successfully fused with a high degree of efficiency and consistency before the cells lost secretion or were overgrown by non-secreters.
Immunofluorescence assay with live cells enabled us to rapidly detect autologous antibodies against antigens on the cell membrane. We could screen many supernatants of EBV-activated B cells or hybridoma cells with this method and successfully cloned human hybridoma cells secreting autologous IgM antibodies. Isolated autologous monoclonal IgM antibodies showed diverse staining patterns in the live cell immunoflourescence assay. Some of them only stained small, shrunken cells that were later proven to be apoptotic cells by immunofluorescence assay and flow cytometry analysis.
Using Jurkat cells as a model, we investigated that autologous monoclonal IgM antibodies bound to different phases of apoptotic cells. AC32 and AC39 IgMs scarcely bound to the early apoptotic cells (4 h after incubation with staurosporine) but bound well to the late apoptotic cells (10 h after incubation with staurosporine). In contrast, AC34 IgM bound well to both early and late apoptotic cells.
Among the changes on the surface of apoptotic cells that facilitate their recognition, the best characterized is the loss of phopholipid asymmetry and the translocation of phosphatidylserine from the inner to the outer leaflet of the lipid bilayer, which occurs very early during the apoptotic process.(11) Previous studies have shown that externalized PS on the surface of apoptotic cells recognized by extracellular bridging molecules, such as β2 glycoprotein, milk fat globule protein (MCF-E8), protein S, growth arrest-specific 6 (Gas 6), and thrombospondin, and they facilitate interactions between macrophages and apoptotic cells.(3) We estimated the reactivity of autologous IgM antibodies to PS and PC. The results demonstrated that AC34 IgM recognized PS well, which means PS might be a possible molecule recognized by AC34 IgM on the surface of apoptotic cells and AC34 IgM might be a possible candidate of bridging molecules between the PS and phagocytes. We investigated the effects of AC34 IgM on the internalization of apoptotic cells by in vitro phagocytosis assay; Jurkat cells induced early apoptosis by 1 μM staurosporine treatment for 4 h and presented to human monocyte-derived macrophages for engulfment. Interestingly, the phagocytosis of apoptotic cells was increased significantly in the presence of AC34 IgM alone. This finding suggested that AC34 IgM might enhance phagocytosis in a complement-independent process. AC34 may enhance phagocytosis of early apoptotic cells by binding PS in apoptotic cells and linking them to the macrophages through the Fc receptors.
Previous studies have shown that the antigen recognized by serum IgM was the PC moiety on lysophosphatidylcholine (LPC), which is formed by hydrolysis of plasma membrane phospholipids and is exposed on the late phase apoptotic cells.(5) Ikeda and colleagues also isolated mouse poly-reactive natural IgM (3B4), which recognizes LPC and facilitates internalization of the late apoptotic cells.(8) In the present study, however, we identified human monoclonal IgM (AC34) recognizing PS antigen that is exposed on the early phase apoptotic cells and AC34 IgM enhanced uptake of early apoptotic cells. This AC34 IgM may facilitate the clearance of early apoptotic cells and prevent the appearance of late apoptotic cells, which are the sources of auto-antigens in autoimmune disorders like SLE. In order to characterize the origin and molecular structure of AC34, the nucleotide sequence of the variable regions of AC34 was examined by RT-PCR and sequencing. The results were aligned to germline gene sequence database and analyzed by IMGT/V-QUEST. Both the VH and VL genes shared high nucleotide identities of 100% and 98%, respectively, with their corresponding germline genes. These genes contained a minimum number of somatic mutational events, suggesting that AC34 was derived from rarely mutated or unmutated germline genes. As to junction regions of H and L chains, nucleotide additions exist at the VH-DH and DH-JH junctions as well as at the VK-JK junction. But deletion mutations of AC34 were not observed.
In conclusion, we obtained an autologous natural IgM AC34 via human hybridoma technique. AC34 could recognize PS on the surface of early apoptotic cells and stimulate the uptake of apoptotic cells by macrophages. The strong V gene homologies indicated that AC34 expresses VH and VK germline genes.
Footnotes
Acknowledgment
This study was supported by Korean R&D cluster grant from Ministry of Knowledge Economy.