
Abstract
Plumbagin (PL; 5-hydroxy-2-methyl-1, 4-naphthoquinone) is an important secondary metabolite, mainly produced in the Plumbago zeylanica L. (Plumbaginaceae). A single-chain variable fragment (scFv) antibody, fusion of the variable regions of the heavy chain and light chain of immunogloblin against PL (PL-scFv) was expressed by Bac-to-Bac Baculovirus Expression System using Spodoptera frugiperda (Sf9) insect cells and characterized to investigate potential use of PL-scFv as a tool for plant immunomodulation. Functional PL-scFv expressed in the Sf9 insect cells were purified using cation exchange chromatography followed by immobilized metal ion affinity chromatography (IMAC). The yields of the purified PL-scFv in the culture supernatant and Sf9 insect cells were 2.0 mg and 5.2 mg per 1 liter of Sf9 culture medium, respectively. Recombinant purified PL-scFv was then characterized by the indirect competitive enzyme-linked immunosorbent assay (ELISA). The cross-reactivity and sensitivity of PL-scFv expressed in Sf9 insect cells were compared with PL-scFv expressed in Escherichia coli and its parental anti-plumbagin monoclonal antibody (MAb 3A3) secreted from hybridoma cells. Intriguingly, the specificity of the PL-scFv expressed in Sf9 insect cells was found to be different from that expressed in E. coli and parental MAb 3A3, although the detectable level (0.2–25 μg/mL) was the same in ELISA using each antibody. Even more interestingly, the characteristics of PL-scFv, which have wide cross-reactivity against 1,4-napththoquinone, suggest its potential use as a tool for plant immunomodulation not only for breeding Plumbaginacea family containing PL but also for breeding other medicinal plants containing bioactive naphthoquinones.
Introduction
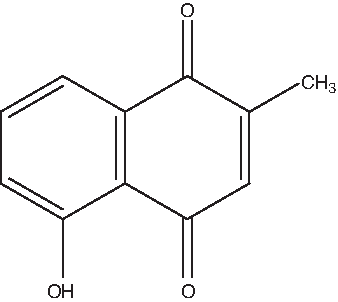
Structure of plumbagin.
Plant immunomodulation is a means to modulate its function by expression of specific recombinant antibody in plant via an antigen-antibody reaction, which was first reported by Owen and colleagues.(12) Until now the target antigen has been expanded to include endogenous small molecules, such as phytohormones, pesticide, and herbicide, to confer its tolerance.(13–15) Furthermore, Putalun and colleagues reported successful immunomodulation of solasodine glycoside by expression of anti-solasodine glycoside single-chain variable fragment (scFv) that stimulates the biosynthesis pathway in Solanum khasianum, suggesting the potential use of scFv as a tool to breed a medicinal plant facing global extinction as a result of overexploitation.(16)
In a previous report, we produced a monoclonal antibody against PL and applied it to the development of an enzyme-linked immunosorbent assay (ELISA) for determination of PL in a PL containing plant.(17) We then constructed, expressed, and characterized an scFv antibody, a variable region of heavy and light chains containing a flexible peptide (Gly4Ser)3, against PL (PL-scFv; DDBJ accession no. AB470492) in Escherichia coli to use it as a tool for immunomodulation.(18) Although PL-scFv expressed in E. coli (PL-scFv [E. coli]) was well characterized by indirect competitive ELISA, there is a possibility that PL-scFv expressed in a bacterial system has different characteristics from that expressed in a plant system. In order to investigate the characteristics of PL-scFv in vitro more accurately before performing time-consuming plant immunomodulation, PL-scFv was expressed in Spodoptera frugiperda (Sf9) insect cells, which provide post-translational modifications similar to those in the plant system, but unlike those in the bacterial system, and characterized by indirect competitive ELISA.
In this baculovirus-insect cell expression system, we constructed a baculovirus donor vector containing honeybee melittin signal sequence (HMSS)(19) to accelerate secretion of the recombinant PL-scFv into Sf9 culture medium. Expression, purification, and characterization of PL-scFv in Sf9 insect cells are demonstrated in this article.
Materials and Methods
Chemicals and immunochemicals
PL was purchased from Wako Pure Chemicals Industries (Osaka, Japan). Ovalbumin and monoclonal anti-T7-tag antibody produced in mouse were purchased from Sigma-Aldrich (Steinheim, Germany). Horseradish peroxidase (HRP)-labeled anti-T7-tag conjugates and Cellfectin reagents were obtained from Invitrogen (San Diego, CA). HRP-labeled anti-mouse IgG goat antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). DNA polymerase and DNA restriction enzyme were purchased from Takara (Kyoto, Japan). All other chemicals were standard commercial products of an analytical reagent grade.
Cell lines and cell culture
Sf9 insect cells line (ATCC no. CRL-1711), derived from the ovarian tissue of Spodoptera frugiperda, and Sf-900 II SFM, a serum-free medium optimized for growth of the Sf9 insect cells, were obtained from Invitrogen. Sf9 insect cells were routinely grown in Sf-900 II SFM medium in shaker flasks at 130 rpm and 25°C. The cells were passaged every 4 days at a seeding density of 5.0 × 105 cells/mL.
Preparation of PL-ovalbumin conjugates
The synthesis of PL-ovalbumin (PL-Ova) conjugates was performed as described in our previous study with some modifications.(17) 1-ethyl-3-(3'-dimethylaminopropyl)-carbodiimide hydrochloride (EDC; 6 mg, 0.031 mM) and 3'-(5-hydroxy-2-methyl-1,4-naphthoquinone-3-yl) propanoic acid (3 mg, 0.012 mM) were added to a mixture of 35% (w/w) pyridine solution (0.3 mL) and MES buffer consisting of 0.1 M 2-(N-morpholino) ethanesulfonic acid and 0.9% (w/v) sodium chloride (0.3 mL). The reaction mixture was added dropwise to 0.3 mL MES buffer containing 3 mg ovalbumin and then stirred at room temperature for 13 h. Subsequently, the mixture was centrifuged, and the supernatants were dialyzed against five batches of H2O for 2 days at 4°C and lyophilized to yield 1.0 mg of PL-Ova conjugate, which was used as a coating antigen in ELISA.
Construction of a baculovirus donor vector
The HMSS peptide used to promote the secretion of PL-scFv into the culture supernatant was amplified by the polymerase chain reaction (PCR) from pMelBac A vector (Invitrogen) using an HMSS specific primer containing BamH I and EcoR I restriction enzyme sites. It was then digested with the same restriction enzyme and ligated into downstream of the polyhedrin promoter of the pFastBac 1 vector (Invitrogen) to generate the pFastBacMel (pFBM) vector. The PL-scFv domain was amplified by fusing it with N-terminus His6-tag and T7-tag domains from the pET28a vector, encoding the PL-scFv gene by PCR using a His6-tag specific primer and a PL-scFv specific primer containing Sal I and Not I restriction enzyme sites, respectively. It was then digested and ligated into the pFBM vector to generate the pFBM/PL-scFv donor vector.
The primers used for the construction of the baculovirus expression vector were as follows: forward primer for HMSS: 5′-CGC
Transposition of the donor vector and transfection of Sf9 insect cells
DH10Bac competent cells (Invitrogen) were used for the production of the recombinant bacmids used in the Bac-to-Bac Baculovirus Expression System (Invitrogen). The DH10Bac E. coli strain contains a parent bacmid that recombines with the donor plasmid pFBM/PL-scFv to create an expression bacmid.
The constructed baculovirus donor plasmid pFBM/PL-scFv was extracted and transformed into DH10Bac to produce a recombinant bacmid by in vivo transposition. Sf9 cells (9.0 × 105 cells) cultured in 35-mm dish were transfected with the recombinant bacmid using Cellfectin reagent (Invitrogen). At 72 h after transfection, the recombinant baculoviruses were harvested by centrifugation at 5000 rpm for 5 min at 4°C from the cell supernatants, and viral titer was determined by plaque assay.(20)
Plaque assay
Viral titer was determined by a plaque assay. The Sf9 cells suspension at 5.0 × 105 cells/mL (12 mL) in Sf-900 II SFM were prepared, and 2 mL of cell suspension aliquoted into each well of 6-well plates. The cells were then settled to the bottom of the plate and incubated at 25°C for 1 h. After removal of supernatant from the plate, 6-log serial dilution (10–2 to 10–7) of the clarified baculoviral stock in Sf-900 II SFM and 1 mL of the appropriate virus dilution were aliquoted into 6-well plates, and then incubated for 1 h at 25°C. Following the 1 h incubation, the media containing virus were removed from the wells and replaced with 2 mL of Sf-900 plaquing medium consisting of 30 mL of Sf-900 medium (1.3 × ) and 10 mL of the melted 4% Agarose gel. The plate was then incubated in a 25°C humidified incubator for 7 to 10 days until plaques were visible. The following formula to calculate the titer (plaque forming units (pfu)/mL) was used.
Expression of recombinant PL-scFv in Sf9 cells
One L of Sf9 cells in the logarithmic growth phase (1.0 × 106 cells/mL) were infected with recombinant baculovirus with a titer of 1.0 × 109 pfu/mL at a multiplicity of infection (MOI) of 5 and further incubated at 25°C for 72 h. The cell cultures were collected by centrifugation at 5000 rpm for 10 min at 4°C and diluted with 20 mL of 50-fold concentrated starting buffer (1 M Tris-HCl and 100 mM β-mercaptoethanol [β-ME, pH 7.2]) for purification. The remaining pellets were treated with 50 mL of starting buffer (20 mM Tris-HCl and 2 mM β-ME [pH 7.2]) containing protease inhibitor (20 μg/mL Aprotinin, 20 μg/mL E-64, 20 μg/mL Pepstatin, and 174 μg/mL phenylmethylsulfonyl fluoride). It was then ultrasonically lysated to extract intracellular PL-scFv of Sf9 insect cells.
Recombinant PL-scFv were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis and characterized by indirect competitive ELISA.
Purification of PL-scFv protein
PL-scFv protein containing the N-terminal His6-tag was purified by cation exchange chromatography using Toyopearl CM-650M (Tosoh Corp., Tokyo, Japan), followed by IMAC using His-bind resin (Novagen, Madison, WI).
First, 10 mL of CM-650M cation exchanger were packed into a column (1.1 × 23 cm) and equilibrated with starting buffer (20 mM Tris-HCl and 2 mM β-ME [pH 7.2]). Then, the samples (1 L of culture supernatant and 50 mL of intracellular fraction) treated with starting buffer were filtrated using a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Milipore, Billerica, MA) and were applied to a cation exchanger. After washing the column with starting buffer to remove unadsorbed proteins, the bound proteins were eluted with a continuous gradient of NaCl from 0 to 1 M in starting buffer. Direct ELISA was carried out to identify the presence of PL-scFv in the fractionated samples.
Subsequently, 6 mL of His-bind resin were packed into the column (1.1 × 23 cm) and charged with 50 mM NiSO4 in binding buffer (50 mM Tris-HCl, 500 mM NaCl, and 20 mM imidazole [pH 7.7]). The positive fractions detected in direct ELISA (20 mL each) were collected and adjusted so that they had the same constitution as the binding buffer using 8-fold concentrated binding buffer (400 mM Tris-HCl, 4 M NaCl, and 160 mM imidazole [pH 7.7]) before being applied to the resin column equilibrated with binding buffer. These samples were then applied on a resin column and washed with binding buffer followed by washing buffer (50 mM Tris-HCl, 500 mM NaCl, and 60 mM imidazole [pH 7.7]) to remove nonspecifically bound proteins. The samples were then eluted with elution buffer (50 mM Tri-HCl, 500 mM NaCl, and 500 mM imidazole [pH 7.7]) and analyzed by direct ELISA and SDS-PAGE. The yield of purified PL-scFv was determined according to the method of Bradford.(21)
Direct ELISA
Direct ELISA was carried out to identify the presence of PL-scFv in the fractionated samples. A 96-well immunoplate (Nunc, Roskilde, Denmark) was coated with PL-Ova (1 μg/mL) conjugates in 100 μL of 50 mM carbonate buffer (pH 9.0) and incubated at 37°C for 1 h. The plate was washed three times with phosphate-buffered saline containing 0.05% (v/v) Tween-20 (PBS-T) and was then treated with phosphate-buffered saline 300 μL containing 10% (w/v) skimmed milk (PBS-sm) for 1 h to reduce non-specific adsorption. Subsequently, 100 μL eluted samples from each test tube were incubated for 1 h. The plate was washed three times with PBS-T and incubated with 100 μL of a 5000-fold diluted solution of HRP-labeled anti-T7-tag conjugate (Invitrogen) for 1 h. After washing the plate three times with PBS-T, 100 μL of substrate solution (0.3 mg of ABTS in 100 mM citrate buffer containing 0.003% [v/v] H2O2) were added to each well and incubated for 15 min. Absorbance was then measured at 405 nm with a microplate reader (Immuno Mini NJ-2300, Nalge Nunc International, Roskilde, Denmark). The positive fractions, with absorbance at 405 nm, were more than 0.200 and were collected for further purification or protein analysis.
SDS-PAGE and Western blot analysis
SDS-PAGE and Western blot analysis were performed according to the methods of Laemmli(22) and Towbin and colleagues,(23) respectively. Briefly, protein samples were separated by 12.5% SDS-PAGE under reducing conditions and then transferred electrophoretically onto a PVDF membrane (Millipore) at 100 V, 90 mA for 3 h in an ice water bath. The membrane was immersed in PBS-sm for 1 h at room temperature to reduce non-specific adsorption. Then it was immersed with 1000-fold diluted solution of anti-T7-tag monoclonal antibody produced by mouse (Sigma) as a primary antibody followed by 500-fold diluted solution of HRP-labeled anti-mouse IgG goat antibody (Santa Cruz Biotechnology) as a secondary antibody to detect N-terminal T7-tag of purified PL-scFv. The binding of HRP-labeled anti-mouse IgG goat antibody was visualized by the addition of substrate solution (1 mg/mL 4-Chrolo-1-naphthol in phosphate-buffered saline [PBS] containing 0.003% [v/v] H2O2).
Indirect competitive ELISA using recombinant PL-scFv
The reactivity of purified PL-scFv antibody was determined in an indirect competitive ELISA. The same procedures as used in the direct ELISA were used until the blocking step. After washing the blocked plate three times, various concentrations of PL (50 μL) in 10% (v/v) methanol were incubated with 50 μL of purified PL-scFv solution (1.0 μg/mL) for 1 h to observe the competition between PL and the PL-scFv. After being washed three times, the plate with PBS-T was incubated with 100 μL of a 5000-fold diluted solution of HRP-labeled anti-T7-tag conjugate (Invitrogen) for 1 h. After washing the plate three times with TPBS, 100 μL of substrate solution (0.3 mg of ABTS in 100 mM citrate buffer containing 0.003% [v/v] H2O2) were added to each well and incubated for 15 min. Absorbance was measured at 405 nm with a microplate reader.
The cross-reactivities (CR) of purified PL-scFv antibody against various compounds were calculated using the method of Weiler and Zenk,(24) as follows:
Where A is the absorbance in the presence of the test compound and A0 is the absorbance in the absence of the test compound.
Results and Discussion
Construction of baculovirus donor vector and recombinant baculovirus
The baculovirus donor vector pFBM/PL-scFv was successfully constructed for the expression of PL-scFv in Sf9 insect cells by cloning the HMSS (63 bp) from the pMelBac A vector (Invitrogen) and the PL-scFv (717 bp) gene from the pET-28a vector (Fig. 2). The pFBM/PL-scFv was designed to contain the His6-tag and T7-tag so that it is easy to purify and detect PL-scFv, and the HMSS to accelerate the secretion of recombinant PL-scFv into the culture supernatant. In order to verify that the pFBM/PL-scFv vector had been constructed correctly by PCR, a recombinant bacmid containing the PL-scFv gene was obtained through transposition in E. coli DH10Bac. Sf9 insect cells were transfected with recombinant bacmid DNA using Cellfectin reagent and secreted recombinant baculoviruses were amplified until the titer of the stocks reached 1.0 × 109 pfu/mL.
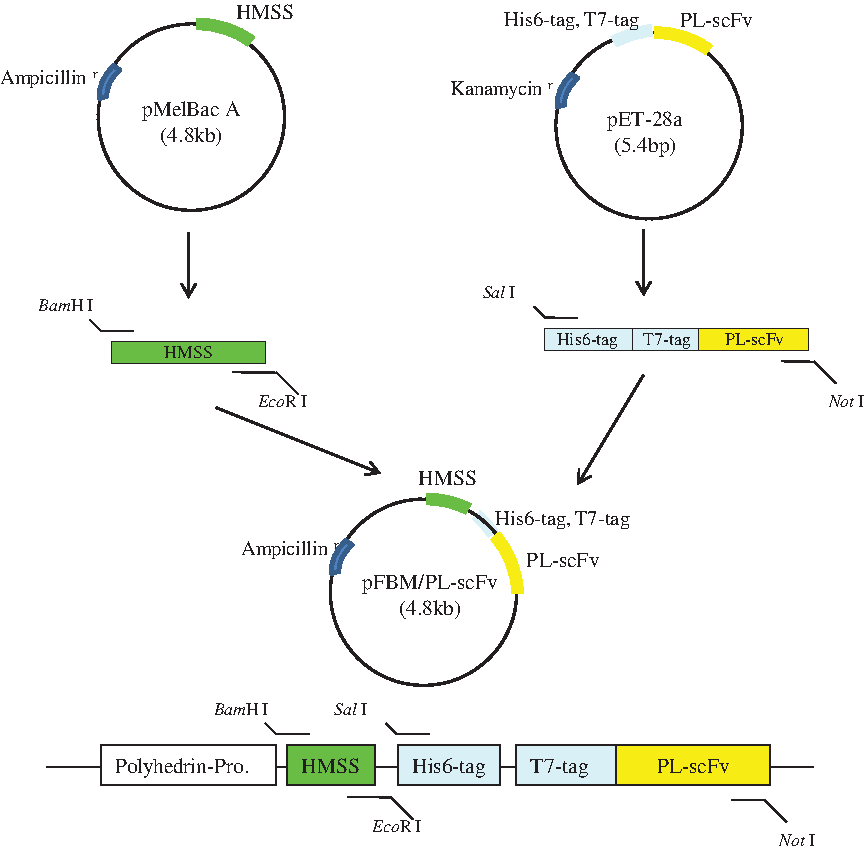
Construction of a donor vector (pFBM/PL-scFv) for expression of PL-scFv in Sf9 insect cells.
Expression and purification of recombinant PL-scFv
Sf9 cells in the logarithmic growth phase (1.0 × 106 cells/mL) were infected with recombinant baculovirus at an MOI of 5 and were further incubated at 25°C for 72 h. Western blot analysis showed that both the culture supernatant and intracellular fraction of Sf9 insect cells infected with recombinant baculovirus exhibited an immunoreactive band with a molecular mass of 29 kDa (Fig. 3). The presence of PL-scFv in the culture supernatant indicated that the HMSS incorporated into the gene construct had successfully targeted the soluble PL-scFv.
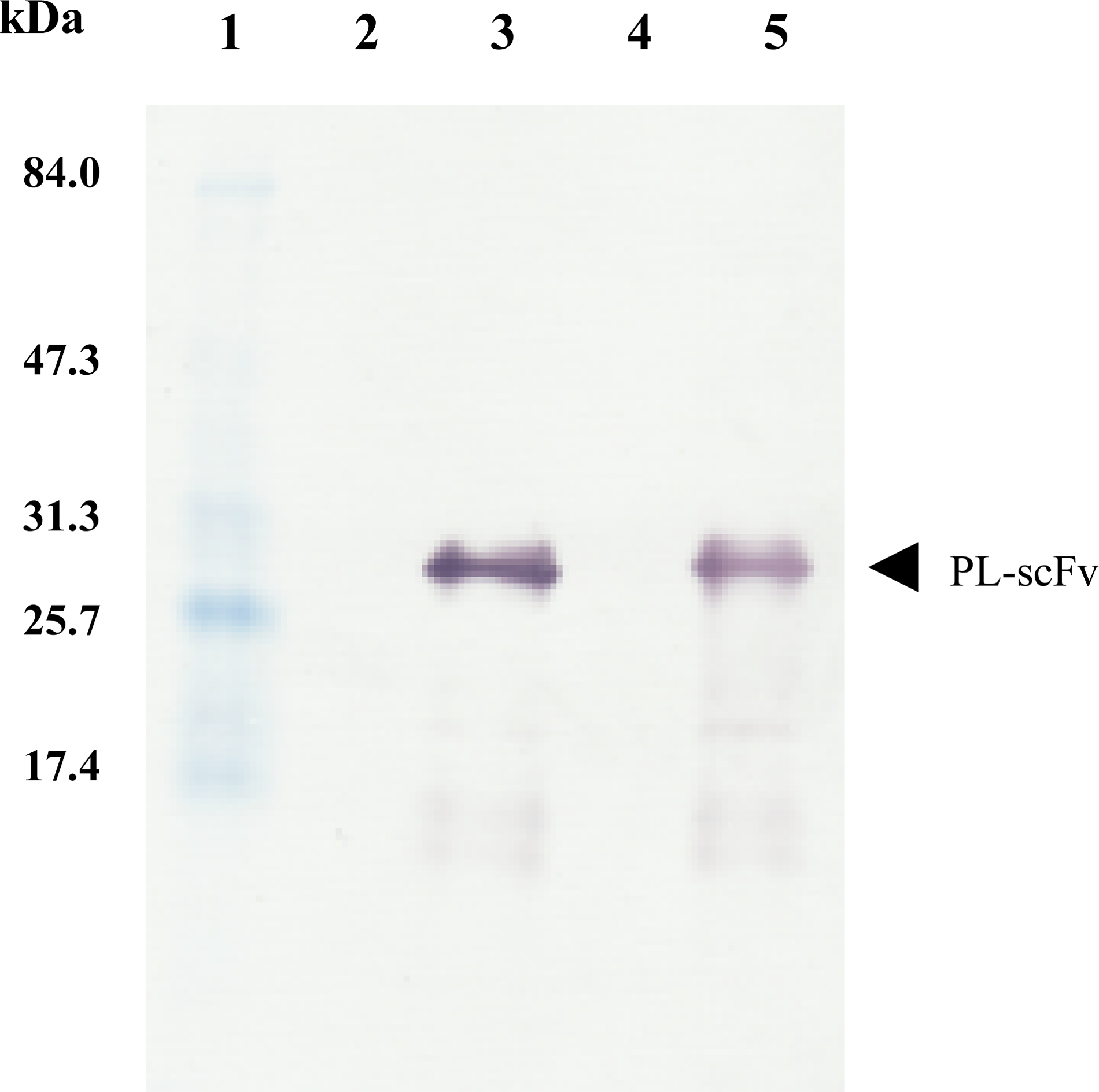
Western blot analysis of PL-scFv produced in intracellular fraction of Sf9 insect cells and in culture supernatant. Intracellular fraction of Sf9 in this protein analysis were prepared by centrifugation at 5000 rpm, 10 min, 4°C, followed by ultrasonic extraction with starting buffer (20 mM Tris-HCl and 2 mM β-ME [pH 7.2]). Its supernatants were 10 times concentrated by Vivaspin 6 (Sartorius Stedim Biotech, Göttingen, Germany), and used as a sample for lanes 4 and 5. Molecular weight marker (lane 1); intracellular fraction of Sf9 before infection with recombinant baculovirus (lane 2); intracellular fraction of Sf9 at 72 h after infection with recombinant baculovirus (lane 3); culture supernatant before infection with recombinant baculovirus (lane 4); culture supernatants at 72 h after infection with recombinant baculovirus (lane 5).
Purification of PL-scFv from the culture supernatant and intracellular fraction of Sf9 insect cells was performed by cation exchange chromatography followed by IMAC. In this purification process, the purity of both PL-scFv purified from the culture supernatant and intracellular fraction were estimated at more than 90% based on Coomassie brilliant blue staining (Fig. 4). The yields of purified PL-scFv in the culture supernatant and the intracellular fraction of Sf9 insect cells were 2.0 mg and 5.2 mg per 1 L of Sf9 culture medium, respectively. Although the yield of PL-scFv expressed in E. coli (21 mg/L) was higher than that expressed in insect cells (7.2 mg/L), the insect cell expression system has advantages over bacterial expression, using it avoids time-consuming refolding and provides post-translational modifications.

SDS-PAGE and Western blot analysis of purified recombinant PL-scFv.
Characterization of recombinant PL-scFv
The purified PL-scFv expressed in intracellular fraction was used for characterization by indirect competitive ELISA because of low yield of PL-scFv expressed into culture supernatant. In this assay, PL-Ova (1.0 μg/mL) was used as a solid-phase antigen. After competition, free PL-scFv (1.0 μg/mL) was bound to a polystyrene micro-immunoplate precoated with PL-Ova. After washing the plate, the amount of PL-scFv antibodies bound to the PL-Ova conjugate was measured using the HRP-labeled secondary antibody against T7-tag and a substrate added to develop color. The detectable range of PL concentrations in this assay was 0.2–25 μg/mL. It became evident from this experiment that the indirect competitive ELISA using PL-scFv expressed in Sf9 insect cells (PL-scFv [Sf9]) displayed the same sensitivity as that using PL-scFv (E. coli) and its parental monoclonal antibody against PL, MAb 3A3 (Fig. 5). To evaluate the specificity of PL-scFv (Sf9), the CR against other compounds was determined using the developed ELISA and the calculation described by Weiler and Zenk.(24) Table 1 shows comparison data of the CR among PL-scFv (Sf9), PL-scFv (E. coli), and MAb 3A3. The specificity of PL-scFv (Sf9) exhibited wider CR than that of PL-scFv (E. coli) and its parental antibody, MAb 3A3, despite PL-scFv having the same nucleotide sequence both in the expression vectors for the bacterial system and Sf9 insect cell system. As we hypothesized in our previous study, the different specificities between PL-scFv (E. coli) and MAb 3A3 might be derived from conformational differences because the conformation of scFv, which does not have a constant region, is altered and different from that of the parental antibody. Therefore, the different specificities of PL-scFv (Sf9) and PL-scFv (E. coli) might occur with slight conformational changes in scFv in the different expression systems. Changes in the post-translational status such as glycosylation, which can occur in the baculovirus system but not in the bacterial system, are probably involved in these differences in specificity. In the amino acid sequence of PL-scFv, Asn62 is present in the consensus sequence of Asn-Xaa-Ser/Thr (Xaa is any amino acid residue except Pro), which usually leads to N-glycosylation.(25) Moreover, there are six threonines that lead to the potential of O-glycosylation in the sequence of PL-scFv as well. Furthermore, the different expression procedures as well as the different purification procedures may have induced slight conformational changes that caused the different specificities. In the bacterial expression, only IMAC was used, while in the case of the baculovirus expression system, a combination of IMAC and cation exchange chromatography was used to purify PL-scFv. In this study, the target of our antibody is a low molecular hapten rather than a protein or peptides, which have high molecular weights. Therefore, slight conformational changes in this antibody would probably cause different specificities between PL-scFv (E. coli) and PL-scFv (Sf9).
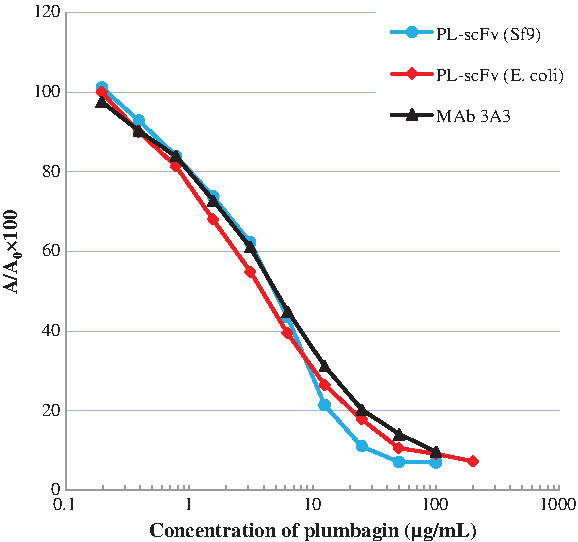
Comparison of calibration curves among PL-scFv (Sf9), PL-scFv (E. coli), and MAb 3A3.The blue circles and blue curve show the standard curve produced using PL-scFv (Sf9). The red squares and red curve show the standard curve produced when PL-scFv (E. coli) were used. The black triangle and black curve show the standard curve produced using MAb 3A3. A/A0, A0 is the absorbance with no PL present; A is the absorbance with PL present.
nd, no data; PL-scFv (Sf9), PL-scFv expressed in Sf9 insect cells; PL-scFv (E. coli), PL-scFv expressed in E. coli; MAb 3A3, monoclonal antibody against PL.
Since single-chain variable fragment against plant secondary metabolites have the possibility of being a powerful tool in breeding a medicinal plant facing global extinction by expression of these scFv that stimulate a biosynthesis pathway in the host plant, PL-scFv having wide CR against PL (5-hydroxy-2-methyl-1,4-naphthoquinone), juglone (5-hydroxy-1,4-naphthoquinone), shikonin ((+)-5,8-dihydroxy-2-(1-hydroxy-4-methyl-3-pentenyl)-1,4-naphthoquinone), and 1,4-naphthoquinone could be an extremely useful tool as well. It is estimated that the specificity of PL-scFv in plants that provide post-translational modifications may behave more like PL-scFv (Sf9) than PL-scFv (E. coli). Therefore, several kinds of bioactive 1,4-naphthoquinones, which react with PL-scFv, could be produced in immunomodulated plants by taking advantage of the wide CR of the PL-scFv (e.g., PL in Plumbago zeylanica L., juglone in Juglans nigra L., and shikonin in Lithospermum erythrorhizon).
In conclusion, we have successfully constructed a donor vector (pFBM/PL-scFv) containing a HMSS to express PL-scFv in a Bac-to-Bac Baculovirus Expression System. Although the total expression level of PL-scFv in the baculovirus system was lower than that in bacterial systems, the baculovirus expression system presented here has advantages over bacterial systems because PL-scFv can be expressed as a functional protein after post-translational modifications unlike the bacterial system. In addition, the specificity of PL-scFv in plants could be estimated using the PL-scFv (Sf9) more accurately than PL-scFv (E. coli). PL-scFv shows the potential to be an extremely powerful tool to use in plant immunomodulation.
Footnotes
Author Disclosure Statement
The authors have no financial conflicts to declare.
Acknowledgments
This work was funded by the Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists. The research in this report was also supported, in part, by a Grant in Aid from the Japan Society for the Promotion of Science Asian CORE Program; the Ministry of Education, Culture, Sports, Science and Technology of Japan; and the National Center for Genetic and Biotechnology (BIOTEC), Thailand.