
Abstract
Peste des petits ruminant (PPR) is an acute, febrile, viral disease of small ruminants with great economic importance. PPR and rinderpest (RP) viruses are antigenically related and need to be differentiated serologically. The use of monoclonal antibodies (MAbs) in ELISA for specific diagnostics and separation of PPR and RPV is important. For this purpose, six Balb/c mice were immunized with inactivated antigen from the Nijeria strain. Fusion cloning was performed 3 months later by directly using cloning plates, selecting the hybridoma colonies at an early stage with an inverted microscope, and transferring the colonies into 96-well plates with a micropipette. From 300 wells, nearly 56 hybridoma clones were found, from which, after testing in ELISA, 11 with higher titer were selected. Among these, only two clones were placed for limiting dilution (1H1, 6A12). Only one clone (6A12L1F12) had no cross-reactivity with RP, reacted with the N protein, and was of IgG2 isotype.
Introduction
Materials and Methods
Reference virus
PPR virus ac.Nigeria 75/1 (EMVT) was used after two passages in lamb kidney and 10 passages in Vero cells. The MAbs were prepared using this virus strain as immunizing antigen and the RPV ac.strain TC Plowright was used after three passages in Razi bovine kidney cells (RBK). This work was done in the quality control department of Razi Vaccine and Serum Research Institute–Iran.
Preparation of viral antigens
Semi-purified viral antigens were prepared from the supernatant of RPV or PPRV-infected bovine kidney culture by the method described,(6) with some modifications. In brief, the infected cells were harvested when showing about 80% cytopathic effect (CPE) and the culture was freeze-thawed three times to release the intracellular virus in culture fluid. Cell debris was removed after centrifugation at 2500 rpm for 25 min (Hettich, East Westphalia-Lippe, Germany). The virus in the supernatant was concentrated by centrifugation at 90,000 g for 2 h (Sorvall Ultra, Wilmington, DE). Then concentrated virus was centrifuged at 90,000 g over a 30% sucrose cushion. The resulting viral pellet was suspended in 1:50 volume of 0.01 M phosphate-buffered saline (PBS). Finally the total protein concentration of antigen preparation was measured using the method described by Lowry and colleagues.(7)
Production of monoclonal antibodies
The monoclonal antibodies used in this study came from several fusions for which 12 Balb/c mice were immunized using crude PPR virus antigen. The immunizing dose was adjusted at a virus concentration equivalent to 3 × 106 infected Vero cells. Three intraperitoneal inoculations were made: the first (day 0) with Freund's complete adjuvant (Sigma, St. Louis, MO), the second (day 35) and third (day 42) with Freund's incomplete adjuvant (Sigma). Subsequently, three immunizations were carried out intravenously without adjuvant on three consecutive days (days 49, 50, and 51) prior to fusion (day 52). However, before fusion, the mice were test-bled on the 45th day of the immunization schedule to detect immune response against the inoculated antigen by indirect ELISA. Spleen cells were fused with mouse myeloma cells, SP2/0 (Razi Institute, Iran), following conventional procedures.(5) Cells were seeded in 96-well microtiter plates (Nunc, Roskilde, Denmark). Well-to-well screening of hybridoma clones, employing an indirect ELISA, was performed at the appropriate stage of growth (25% of the well surface covered by cells). Positive clones were amplified, followed by single-cell cloning and subcloning of PPR-positive hybridoma using the limiting-dilution method.(8) Single-cell cloning was repeated at least twice to prove the monoclonality of the hybridoma culture supernatants.
Selection of specific MAb for PPR
A panel of monoclonal antibodies (MAbs) produced against PPR was screened for specificity by testing against both RP and PPR virus antigens in an indirect ELISA. The MAb showing maximum specificity towards PPR was selected.
The selected MAb was studied for its binding to N protein in a Western blotting semi-dry system (Bio-Rad, Hercules, CA).
Indirect ELISA
Maxisorp ELISA plates (Nunc) were coated with semi purified RPV and PPRV at 100 μL/well containing 2.5 μg/mL in carbonate bicarbonate buffer (pH 9.6, Fluka, Buchs, Switzerland) and incubated overnight at 4°C. The antigen concentrations were established based on experience with PPRV in stage of selection of MAbs. Following four washes in PBS containing 0.05% Tween-20 (PBST, Merck, Darmstadt, Germany) and treatment by blocking buffer (5% skim milk [Sigma] in PBST), the supernatant of eight clones were added and incubated for 1 h at 37°C. The plates were washed and specific binding of antibody was detected with anti-mouse whole immunoglobulin horseradish peroxidase (HRP) conjugate (Sigma) diluted in PBST. The optimal dilution of conjugate (1:15,000) was determined by indirect ELISA. Following the addition of conjugate and incubation for 1 h at 37°C, the reaction was developed using BM Blue (Roche, Mannheim, Germany). Finally the reaction was stopped using 2 N H2SO4 and the wells were read at 450 nm in an ELISA reader (Bio-Rad).
Concentration and isotype determination of monoclonal antibodies
The culture supernatant of high titer positive hybrid was collected and concentrated by ammonium sulfate (Merck) precipitation (50%) and dialyzed (Sigma) against PBS. Their immunoglobulin subclasses were determined by isotyping kit (Roche).
Immunoblotting analysis of antibodies
The purified PPR virus was run on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, Bio-Rad) through 10% vertical slab gel under denaturing conditions.(9) Proteins separated on a polyacrylamide gel were transferred to nitrocellulose membrane (Sigma), according to the procedure of Madani and colleagues,(10) using semi-dry transfer apparatus (Bio-Rad). The blot was then blocked with blocking buffer (5% skim milk in PBST) for 1 h at room temperature and incubated with appropriate dilution of selected MAb (specific for PPR) in dilution buffer (2% skim milk in PBST). After incubation for 1 h at room temperature, anti-mouse whole immunoglobulin HRP conjugate (Sigma) in dilution buffer was added and the blot was incubated for 1 h at room temperature. Finally the blot was developed using chloronaphthol (Fluka) in PBS containing 0.01% H2O2.
Results
When sera samples were tested, there was a significant increase in indirect ELISA signal after immunization of 12 Balb/c mice. The sera titers were checked in which the highest titer of one of them was 1:640, and the same mouse was selected for fusion (Table 1). The supernatant screening of clones were done by the same ELISA method, and 11 clones were positive for PPR virus-specific antibodies on initial screening (Table 2). Primary clones were subjected to single-cell cloning and subcloning. Two single clones 1H1 (1.578) and 6A12 (1.602), due to their higher optical densities (OD), were selected among other clones. For monoclonality, these two clones were also taken for limiting dilution (screening of new selected clones are shown in Table 3). Monoclonality of a clone was accepted only when all the wells of a microtiter plate with growing cells gave a positive reaction in indirect ELISA after repeated subcloning. A panel of six MAbs after limiting dilution was raised against the PPR and was used in the indirect ELISA to assess their cross-reactivity with RPV. All antibodies recognized both viruses except MAb 6A12-1F11, which reacted with PPR only (Fig. 1).
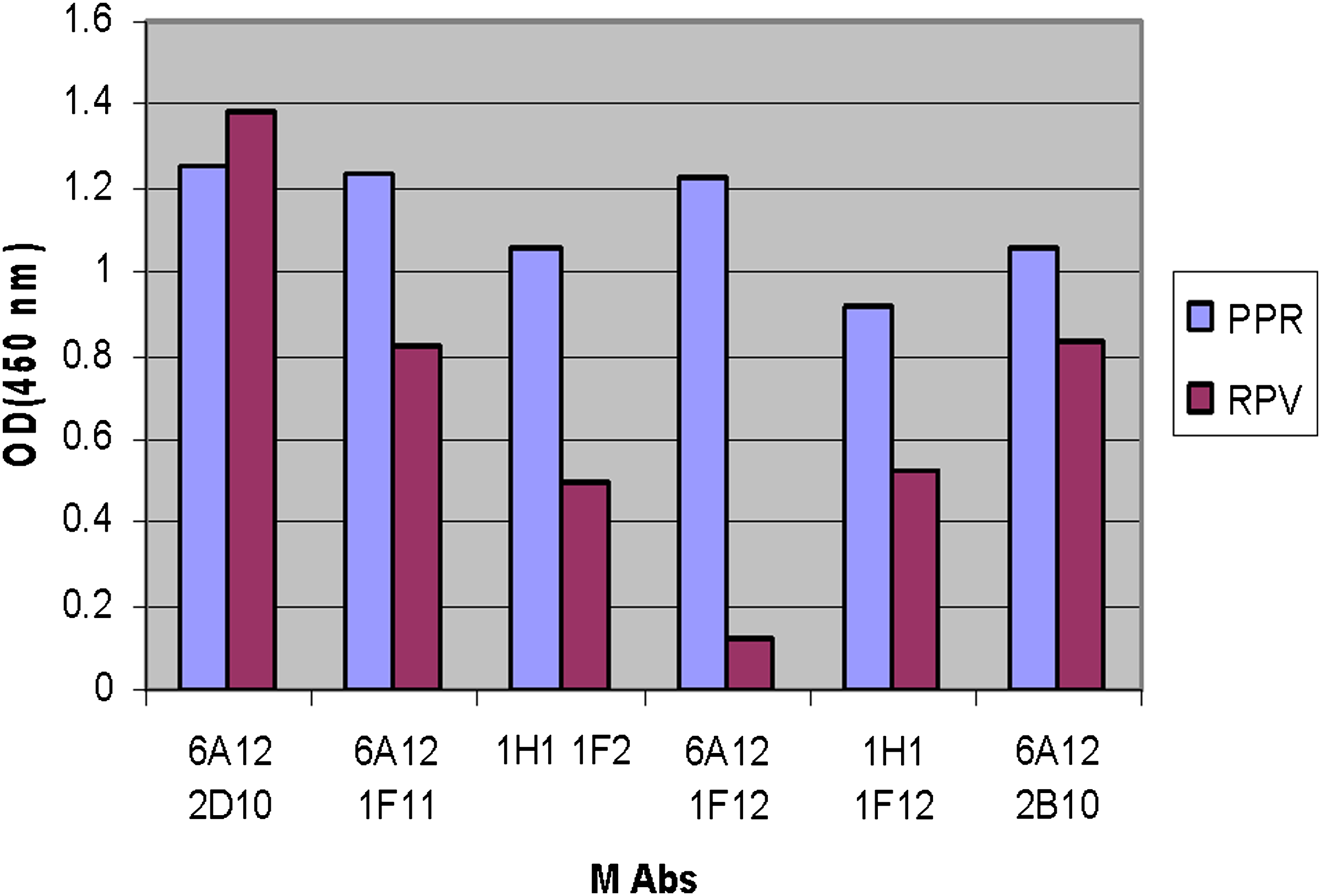
Cross-reactivity between PPR and RPV to show specificity of monoclonal antibody.
The isotyping kit for 6A12-1F11 clone was used and showed a single band on IgG2 class with κ chain (Fig. 2). The immunoblot analysis for 6A12-1F11 clone was examined. Protein bands of the purified virus transferred to nitrocelluse paper, which showed a single and sharp band on N-protein zone with 68 kDa molecular weight. This sharp and single band itself proves the monoclonality of the above clone (Fig. 3) against N-protein of PPRV.
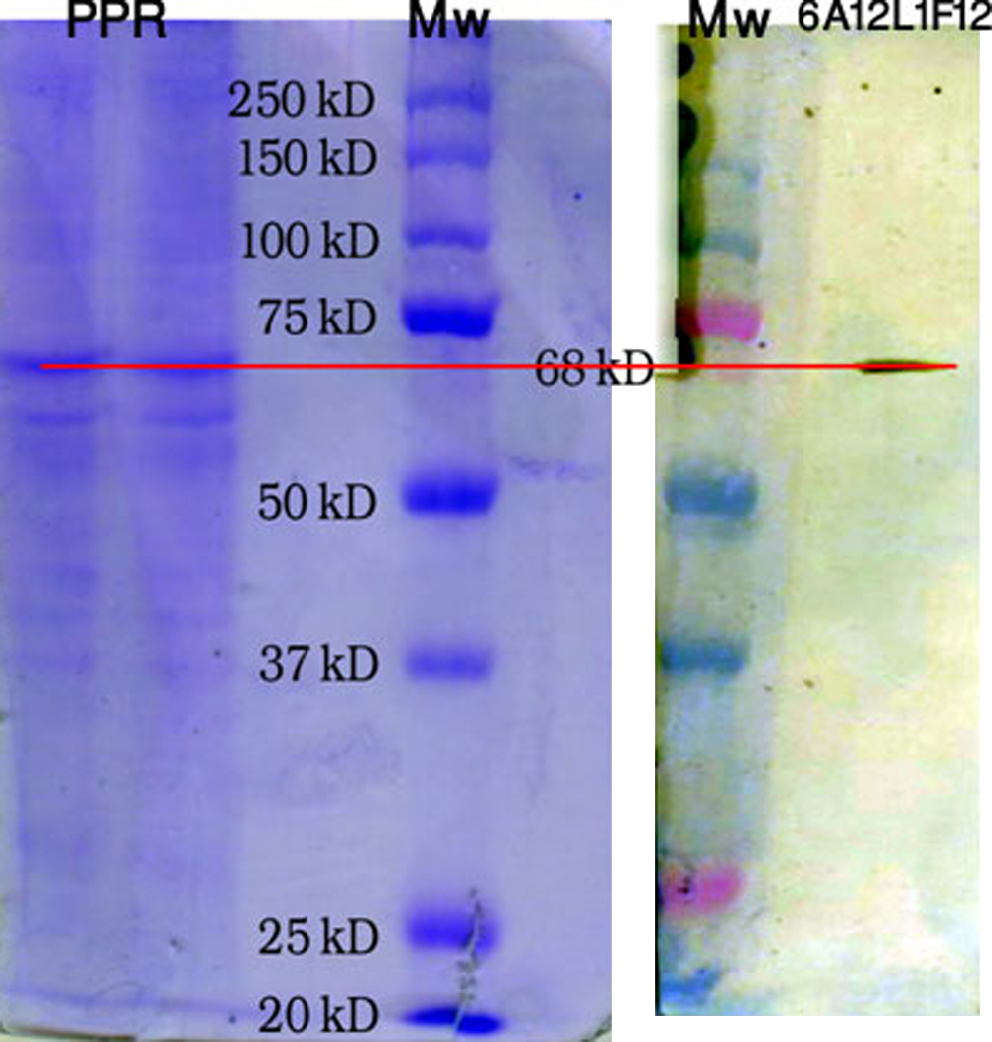
Immunoreactivity of MAb 6A12 L1F12 against N protein of PPR. PPR proteins were separated on a 10% acrylamide slab gel by SDS-PAGE and transferred to a nitrocellulose membrane. The product was immunostained with MAb 6A12 L1F12.

Result of isotyping strip with MAb 6A12 L1F12.
Discussion
Peste des petits ruminants are a highly contagious and economically debilitating viral disease affecting goats, sheep, and wild ruminants. The disease is currently circulating in Asian and African countries, creating problems in small ruminant farming.(2) The N protein is the major viral protein in morbilliviruses. It folds and protects the genome.(3) Among the structural proteins of morbilliviruses, the N protein is the highly conserved immunogenic core protein. Furthermore, the N protein has both type-specific and cross-reactive epitopes. Therefore, it is a good candidate antigen for diagnosis of morbilliviruses.(11)
The monoclonal antibody in this study reacted with the N protein and was of IgG2 isotype. The supernatant of this clone was also tested in hemagglutination inhibition and a virus neutralization test and was found to be negative (data not shown). This work produced findings similar to other research results, which are provided as follows. It has been suggested by Dhinakar and colleagues(3) that the N-protein of some RP viruses is phosphorylated and hence migrates as a doublet or triplet. The same could be true for PPR virus isolates since the N-proteins (precipitated by the MAbs) of some of the isolates were seen as doublets. The slower migrating band could be the phosphorylated form of the N-protein. It is not clear why the N-protein of some strains is phosphorylated while others are not. In 2002 Singh and colleagues(5) reported that anti–N monoclonal antibody (clone 4G6) was found to be useful for specific diagnosis of PPR virus infection from clinical samples using a sandwich ELISA. N-protein of morbilliviruses has always been the focus of attention for differentiation of closely related viruses. The main advantage of using N-protein–based diagnostics is the abundance of this protein in clinical materials(12,13) and the availability of specific as well as cross-reacting epitopes.(4) In this protein the cross-reacting epitopes help capture the antigen through antibodies directed to common epitopes, while specific epitopes help in the detection of viruses using PPR-specific monoclonal antibodies. In 2004 Singh and colleagues(5) reported that 23 MAbs were produced using a semi-purified preparation of an Indian virus isolate (PPR sungri 96) as immunogen. In the majority of MAbs at least 14 precipitated internal virion proteins (nucleoprotein and matrix protein) in RIPA. Singh also observed that anti-N-protein antibodies co-precipitated actin (DNA binding protein) along with viral nucleoprotein, and these are the major background host proteins. This suggests that these viruses do not shut off host cell protein efficiently.
The MAbs produced are specific to PPR viruses and do not react with the vaccine strain of RP virus. They will be highly useful in the differential diagnosis of PPR from RP. This MAb will also be useful for measurement of PPR virus–specific antibodies in a competitive ELISA although these titers may not correlate with protective titers and those against the H protein.
Author Disclosure Statement
The authors have no financial conflict to disclose.