
Abstract
Ferric uptake regulator A of Mycobacterium tuberculosis (MTB), which belongs to the Fur superfamily, is situated immediately upstream of katG encoding catalase-peroxidase, a major virulence factor that also activates the pro-drug isoniazid. The feature and role of FurA in oxidative stress contribute to research on the pathogenesis of mycobacteria. In this study, four novel mouse monoclonal antibodies were generated using the prokaryotically expressed FurA protein as immunogen. The furA gene of M. tuberculosis H37Rv was inserted into a bacterial expression vector of pRSET-A and effectively expressed in Escherichia coli BL21(DE3). The expressed fusion protein existed as soluble form in cell lysates and was purified via Ni-NTA purification system. Using the fusion protein to immunize BALB/c mice, four monoclonal antibodies (H9H6, H9E12, H10H6, and H10H8) were produced. As shown by Western blot analysis and cell fluorescence microscopy assay, the four antibodies could recognize the FurA protein, respectively. Then we assessed the effect of iron on the expression of FurA in MTB H37Rv and we concluded that iron does not affect FurA expression. These results suggest that the antibodies against FurA may provide a powerful tool for elucidating FurA biofunctions and regulation mechanism in the pathogenesis of tuberculosis.
Introduction
In enteric bacteria, the oxidative stress response is mediated by the regulated expression of katG and aphC in addition to other factors.(15) Absence of functional components of the oxidative stress response appears to be a common theme in pathogenic mycobacteria, albeit such phenomena appear counterintuitive in the context of intracellular survival in infected macrophages. The pathogenic mycobacteria have a limited, non-protective oxidative stress response, which in the case of MTB is restricted to induction of a single gene, katG, encoding the catalase-peroxidase.(16,17) In parallel to the potential role in pathogenicity, KatG has been shown to participate in acquired resistance and innate susceptibility to isonicotinic acid hydrazide.(16,18,19) Indeed, inactivation of katG is frequently found among INH-resistant MTB strains.(20)
Apart from oxyR-dependent regulation of oxidative stress response, many organisms couple the expression of oxidative stress genes with iron metabolism, principally via the ferric uptake regulator Fur.(21) Fur-like proteins are widespread in both gram-negative and gram-positive bacteria.(22–24) Their major role in the regulation of the iron-uptake system in response to environmental iron was first described for Escherichia coli.(25) Fur is an iron-dependent transcriptional regulator that regulates genes related to iron acquisition, oxidative stress response, basic physiological processes, and various other functions.(26–29) Therefore, Fur has gathered significant interests as a potential target for novel antibiotics.(30)
MTB has two Fur genes: furA and furB.(31) The furA gene is located immediately upstream of the katG gene encoding the catalase-peroxidase enzyme. KatG is involved in the oxidative stress response and a significant virulence factor of MTB. furA-katG region of MTB, encoding a Fur-like protein and the catalase-peroxidase, is highly conserved among mycobacteria. Both genes are induced upon oxidative stress. FurA is co-expressed along with KatG and it auto-represses its expression by binding to a unique sequence upstream of the furA gene.(32) The genetic linkage of furA and katG in all species of mycobacteria has suggested a putative involvement of the FurA protein in katG regulation and isoniazid resistence. Indeed, recent studies have demonstrated that it is a second regulator of oxidative stress response in mycobacteria and that it negatively controls katG. In MTB and M. smegmatis, FurA appears to be a dominant regulator affecting mycobacterial physiology and intracellular survival.(33) Although FurA and FurB belong to the same protein family, they only share a sequence identity of approximately 25%. Thus, while it is likely that they have a similar structure overall, certain key elements in their respective architectures are likely to differ considerably. For instance, some of the distinct structural details discriminate in divalent metal binding while others are critical to the recognition of DNA sequence motifs in the operator of regulated genes. FurB is induced by zinc and is involved in the regulation of zinc homeostasis.(34)
The furA gene has been identified in MTB as well as other mycobacterial species.(32,33,35–37) S. reticuli FurS and MTB FurA show 53% identity, and the predicted secondary structures of the two proteins are similar. Moreover, typical motifs involved in metal binding and several cysteines sense that the oxidative stress are conserved.(38,39) Thus, the two proteins play similar roles in responding to oxidative stress. FurA is of particular biomedical importance as it controls a protein central to contemporary TB therapy. One of the more efficacious therapeutics for this disease is isoniazid (INH), which is able to traverse the complex lipid membrane of MTB, entirely by passive diffusion. While unmodified INH is not toxic to the pathogen, it becomes activated by the mycobacterial catalase KatG, which modifies INH into a range of reactive intermediates, including NAD+ and NADP+ adducts. INH-resistant MTB strains isolated from patients predominantly have mutations in the katG gene leading to a catalase-peroxidase with reduced or abolished catalytic activity. Therefore, FurA has attracted increased interest as a potentially novel drug target for the development of inhibitors that could enhance KatG levels, possibly boosting INH potency.
In order to understand the function of FurA, we cloned, expressed the furA gene of MTB in E. coli, and generated monoclonal antibodies against FurA. Then we assessed the effect of iron on the expression of FurA in MTB H37Rv.
Materials and Methods
Bacterial stains, plasmids, and cells
E. coli DH5α, E. coli BL21(DE3), and MTB H37Rv competent cells were kept in our laboratory. E. coli DH5α and E. coli BL21(DE3) were cultivated in Luria-Bertani (LB) medium. MTB H37Rv was grown at 37°C for 18 days in liquid Middlebrook 7H9 medium (Difco Laboratories, Detroit, MI) supplemented with 0.2% glycerol, 0.05% Tween-80, and 10% ADC enrichment (Becton Dickinson, Frankin Lakes, NJ). The plasmid vector pGEM-T-Easy, pRSET A, and pcDNA3.1(-) were purchased from Invitrogen (Carlsbad, CA). Restriction enzymes were purchased from Takara (Dalian, China). HRP-conjugated goat-anti-Mo IgG polyclonal antibody was from Zhongshan Bio (Beijing, China). RPMI-1640 medium was from Gibco (Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Gibco) at 37°C in a humidified atmosphere. Unless otherwise stated, all laboratory reagents and antibodies were purchased from Sigma (St. Louis, MO).
furA DNA synthesis, cloning, and sequencing
The coding sequence of furA (GenBank accession no. AF002194) was obtained from MTB genomic DNA by polymerase chain reaction (PCR) using the following primers––forward primer: 5′-
Subcloning of furA
To construct the prokaryotic and eukaryotic expression plasmids, pGEM-T-Easy-furA was cleaved at the BamHI/HindIII sites and the furA fragments were purified. The sequenced full-length furA was ligated into the expression vectors pRSET A with N-terminal His tag and pcDNA3.1(-), respectively. Resulting plasmids were checked by DNA sequencing. pRSET A-furA was transformed into E. coli BL21(DE3) to express the FurA protein, while the eukaryotic expression vector pcDNA3.1(-)-furA was amplified in E. coli DH5α and purified for transfection into Cos-7 cells. Restriction endonuclease digestion, ligations, transformation, and plasmid DNA isolation were conducted by standard methods.
Expression and purification of recombinant protein
A single colony of a E. coli BL21(DE3) cells harboring the pRSET-A-furA plasmid was induced and cultured overnight at 37°C in Luria-Bertani (LB) liquid medium containing kanamycin (50 μg/mL), then transferred to fresh medium and incubated for another 2 h until the optical density (OD600) of the cultured cells reached 0.6. Expression was induced by 0.1 mM of isopropyl-β-D-thiogalactopyranoside (IPTG) for 3 h at 33°C. Cells were harvested and resuspended in 1 mL buffer A (50 mM Tris-HCl and 200 mM NaCl [pH 7.5]). The suspension was sonicated on ice 20 times for 10 s with 10 s intervals. Fractions were then analyzed by 12% SDS-PAGE. At the same time, E. coli BL21(DE3) strain harboring the pRSET-A empty vector were induced and analyzed as controls.
For purification of His-tagged protein, the bacterial pellet was resuspended in 20 mL of lysis buffer. The sample was centrifuged at 12,000g at 4°C for 15 min. The recombinant protein was purified using a column with Ni-NTA agarose (Qiagen, Valencia, CA) following the manufacturer's instructions, and was verified by Western blot analysis using anti-His monoclonal antibody (Sigma). The concentration of purified protein was measured using BCA kits (Bios, China).
SDS-PAGE gel electrophoresis and Western blot analysis
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was performed according to Laemmli,(40) using 5% stacking gel and 12% separating gel. After electrophoresis, the proteins were electronically transferred onto nitrocellulose membranes. The membrane was blocked with 5% (w/v) skim milk powder in PBS for 1 h at room temperature followed by incubation with the antibody (MAbs or anti-His MAb) diluted in blocking solution overnight at 4°C. After being washed three times (10 min each) with 0.05% Tween-20 in PBS, the membrane was incubated for 1 h with horseradish peroxidase-conjugated goat anti-mouse IgG at room temperature. The membrane was washed and then analyzed using the enhanced chemiluminescence detection system (Genmed, Boston, MA).
Preparation of the monoclonal antibodies
For the preparation of MAbs, BALB/c mice (6 weeks old) were immunized subcutaneously with 50 μg of the purified protein His6-FurA emulsified in Freund's complete adjuvant (Sigma). Three booster injections were given with 50 μg of the protein emulsified in Freund's incomplete adjuvant at 2-week intervals. Three days after the last injection, the spleen was removed and the spleen cells were fused with Sp2/0-Ag-14 mouse myeloma cells. The culture supernatant from the wells with growing cells was examined for the presence of antibodies to FurA, as described below. The colonies producing anti-FurA antibodies were cloned twice at a density of 0.4 per well by limiting dilution. The cloned cells were injected into the mouse belly cavity and ascitic fluids with anti-FurA MAbs were generated. The culture supernatant from the cloned cells was used to characterize the MAbs. The isotypes of the MAbs were determined with a commercially available isotyping kit (Hyclone Laboratories, Logan, UT). The MAbs were purified using caprylic acid and ammonium sulphate per protocol.
MAb titer determination by ELISA
The titers of MAbs were examined using an indirect enzyme-linked immunosorbent assay (ELISA). The purified FurA protein was dissolved to 10 μg/mL in 50 mM carbonate salt buffer (pH 9.6) and coated on polystyrene microtiter plates (Greiner, Frickenhausen, Germany) at 100 μL aliquot per well overnight at 4°C. After incubation at 4°C, the wells were washed three times (10 min each) with 0.05% Tween-20 in PBS. The coated wells were blocked with 5% (w/v) skim milk powder in PBS for 1 h at room temperature and then washed as previously described. Following this, 150 μL of the culture supernatant from the cloned cells or ascitic fluids with different deliquiation (from 1:10 to 1:108) was added into the wells. After incubation for 2 h at 37°C, the wells were incubated with 150 μL of horseradish peroxidase-conjugated goat anti-mouse IgG (dilution 1:10,000; Sigma) for 1 h at 37°C, then washed. OPD was used as the peroxidase substrate. After 15 min, color development was stopped with 50 μL H2SO4 (2 M) and the absorbance was measured at 490 nm in a Sunrise ELISA Reader (R-Team, Hombrechtikon, Switzerland). Each test was performed in triplicate, and the absorbance at 490 nm of wells without antigen was subtracted from that of wells with antigen before analysis. Antibody titer was defined as the dilution times of the antibody corresponding to the absorbance of 0.500 at 490 nm.
ELISA additivity tests for epitope identification of MAbs against FurA protein
To analyze the antigenic sites recognized by MAbs, ELISA additivity tests were performed. Plates were coated, blocked, and washed as described above. Ascites were diluted at a concentration sufficient to saturate the coated antigen and pairwise added to each well; the remaining steps were performed by indirect ELISA assay. In the ELISA additivity test, the additivity index (AI) value was used to analyze whether MAbs recognize the same antigen site. If two antibodies bind the same site, the AI value will tend towards nil; in contrast, if the two antibodies bind independently at topographically unrelated sites, the AI value will be close to 100.
Transfection of Cos-7 cells and Western blot analysis
The Cos-7 cells were passaged and subcultured on coverslips up to 50–70% confluency. Cells were washed with phosphate-buffered saline, and then 1.5 mL serum-free medium was added to each well. Cos-7 cells were transfected transiently with plasmids pcDNA3.1(-) and pcDNA3.1(-)-furA, respectively, using Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions. After incubation for 6 h, each well was replaced with 2 mL complete medium and was incubated for 48 h. For Western blot analysis, the cells were harvested and were resuspended in lysis buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM b-glycerophosphate, 1 mM Na3VO4) containing proteinase inhibitors.
Fluorescence microscopy
The Cos-7 cells were transfected with the recombinant pcDNA3.1(-)-furA. Cell fluorescence microscopy assay was performed after 48 h of transfection. Cells were grown on coverslips in a six-well plate. After washing with cold PBS, cells were fixed with 2 mL of 4% paraformaldehyde for 20 min. Cells were then washed three times with cold PBS and permeabilized with 2 mL of PBS with 0.1% Triton X-100 for 15 min. After washing four times with cold PBS, cells were incubated with the MAbs H10H6, H10H8, H9E12, and H9H6 supernatant (10 μg/mL, diluted with 1% BSA), respectively, overnight at 4°C. Cells were washed three times with PBS and then incubated with Cy3-conjugated sheep anti-mouse IgG and Hoechst 33258 (Sigma) for 30 min at 37°C. To detect the nuclei, specimens were further incubated with 10 μg/mL of Hoechst 33258 for 5 min. Background staining was removed by washing with PBS three times. The coverslips were mounted on slides and were examined using laser microscope (Nikon, Tokyo, Japan).
Effect of iron on FurA activity
To assess the effect of iron on FurA, a 3-week-old MTB H37Rv culture grown in 7H9 medium with albumin-dextrose-catalase (ADC) supplement was diluted at 1:200 in medium under three conditions for 2 days before preparation of protein analysis: in r7H9 medium (iron-rich group) with a high iron content (5 mM ferric chloride); in regular 7H9 medium (normal group); and in n7H9 medium (no iron group) with 75 μM of dipyridyl (in stock of 200 mM in ethanol). Then MTB H37Rv. After 2 days, the bacterial were collected and prepared for Western blot analysis.
Results
Cloning full-length furA gene and construction of expression vector
Complete sequence of furA gene was amplified from MTB H37Rv. Specific primers were designed according to the published full-length of the furA gene of MTB H37Rv. 525 bp PCR amplification product (Fig. 1) was obtained. The furA gene BamHI and HindIII sites were designed in the primers to facilitate cloning into pRSET A expression vector. The PCR products were first cloned into pGEM-T-Easy vector, which was supplied as linearized with a single 3′-T overhang for TA cloning to improve the efficiency of ligation with PCR products. The BamHI-HindIII fragment was then subcloned in the pRSET A prokaryotic expression vector and pcDNA3.1(-) eukaryotic expression vector, respectively (Fig. 2). The insert sizes were confirmed by digestion and further verified by sequencing.
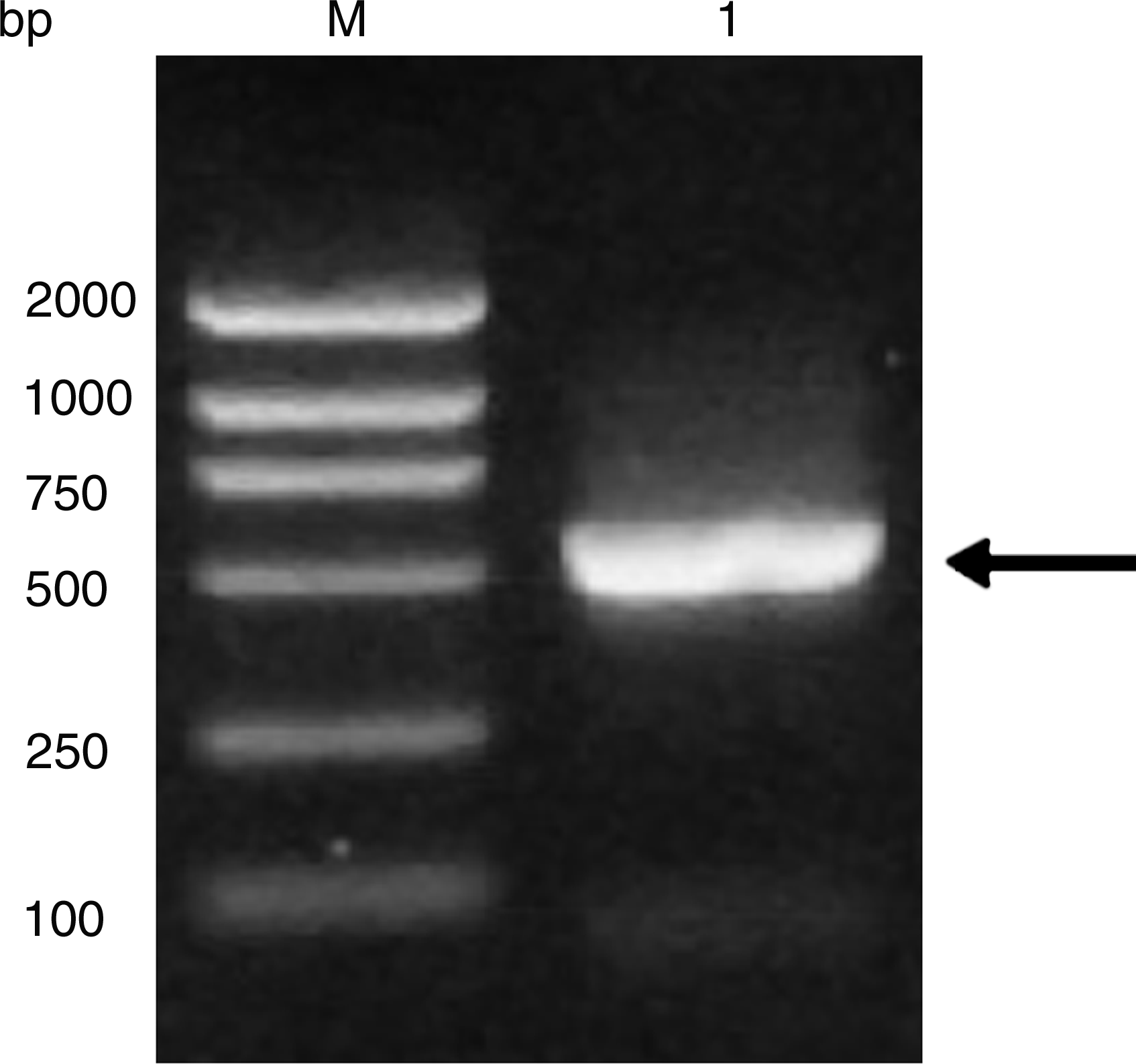
PCR amplification of furA. Lane M, DNA marker DL2000 (Takara); lane 1, PCR product. The specific band is indicated with arrows.

Construction of pRSET A-furA. Schematic of the ORF of furA inserted in the pRSET A expression vector.
Expression, solubility analysis, purification, and identification of FurA protein
Products of the constructs were introduced into an E. coli strain BL21(DE3) cell and expression of the fusion protein was then induced at 37°C. Under the control of IPTG-inducible phage T7 promoter, furA DNA in pRSET A is predicted to encode a recombinant protein of 175 aa with a molecular weight of ∼23 kDa. Small-scale cultures of the positive clones were subjected to IPTG induction to identify clones capable of expressing the predicted recombinant proteins. Recombinant proteins migrated at apparent molecular weight that matched the prediction (Fig. 3). The amount of gene expression reached the maximum 3 h after induction (at 37°C). The results confirmed that FurA could be efficiently expressed in E. coli host cells. Expression was not detected in uninduced recombinant strains. Western blot results confirmed the expressed proteins were the target proteins (Fig. 3).
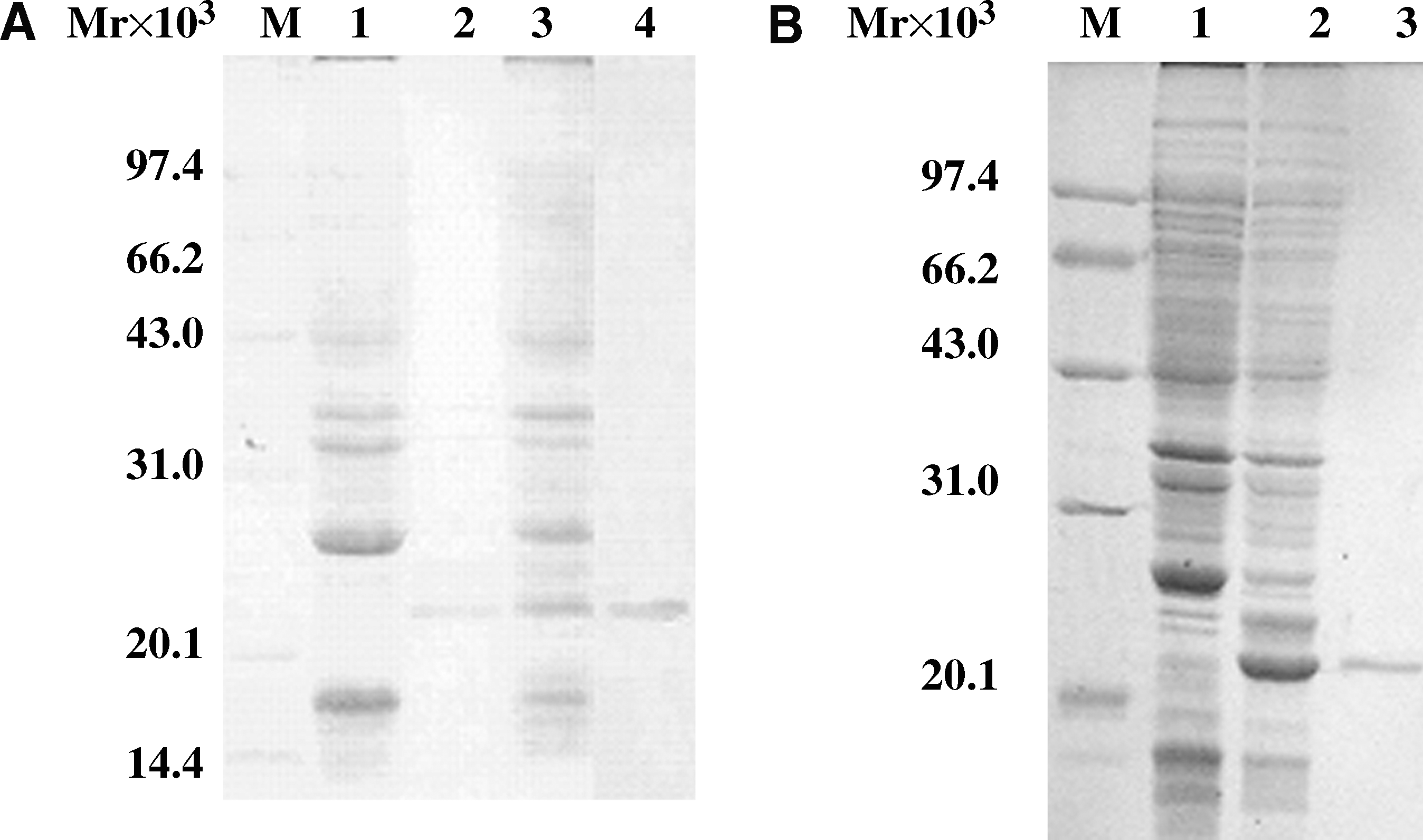
Expression, purification, and Western blot analysis of His6-tagged FurA. (
To examine the relative distribution of the expressed recombinant protein in the soluble and insoluble fractions, both the supernatant and the pellet of the cell lysates were examined to detect the recombinant proteins. The expression of both recombinant proteins was detected predominantly in the soluble fraction (Fig. 3).
The recombinant protein contained a His-tag sequence, which could bind to the Ni-NTA agarose. The target protein was recovered by eluting with imidazole. The His-tagged fusion protein had two fractions, soluble and insoluble. It is important to note, however, that the inducing conditions (temperature, time, and concentration of IPTG) can have a direct effect on the form of the fusion protein. If purification under native conditions is preferred or necessary, the fusion protein must be soluble. Therefore, even though some proteins are insoluble there are generally soluble proteins that can be purified in their native form. The protein used as the antigen must also possess a high degree of purity. To reduce the presence of unspecified proteins that can bind with the Ni-Resin, a low concentration of imidazole was added to the binding buffer.
After purifying the recombinant proteins by affinity chromatography, predominantly one band of the recombinant protein could be seen in the SDS-PAGE and Western blot with a His-tagged monoclonal antibody, indicating that the recombinant proteins were highly purified (Fig. 3). The protein yield was 1.0 mg/mL for FurA.
Characterization of the MAbs
Four mouse MAbs designated H9H6, H9E12, H10H6, and H10H8 were produced using the purified fusion protein His6-FurA, and all four MAbs specifically recognized the His6-FurA fusion protein expressed in E. coli BL21(DE3) by both ELISA and Western blot analysis. All four MAbs reacted with the His6-FurA specifically, while they did not react with the irrelevant protein (His6-FurB). This suggests that MAbs recognize the epitopes on FurA, though not on His6. Titer assay illustrated that the MAbs generated by the H10H6 had the highest titers in both ascitic fluids and purified products (Table 1). It is important to delineate the immunoglobulin class of the four MAbs to determine their potential functional characteristics. The result of subtype analysis showed that MAbs H9H6, H9E12, and H10H6 belonged to IgG1, and H10H8 belonged to IgG2a.
Relative affinity of the MAbs
The relative affinity of the four MAbs was determined by ELISA. The relative affinity of the MAbs was sequenced as H9E12, H10H6, H10H8, and H9H6 (Fig. 4).
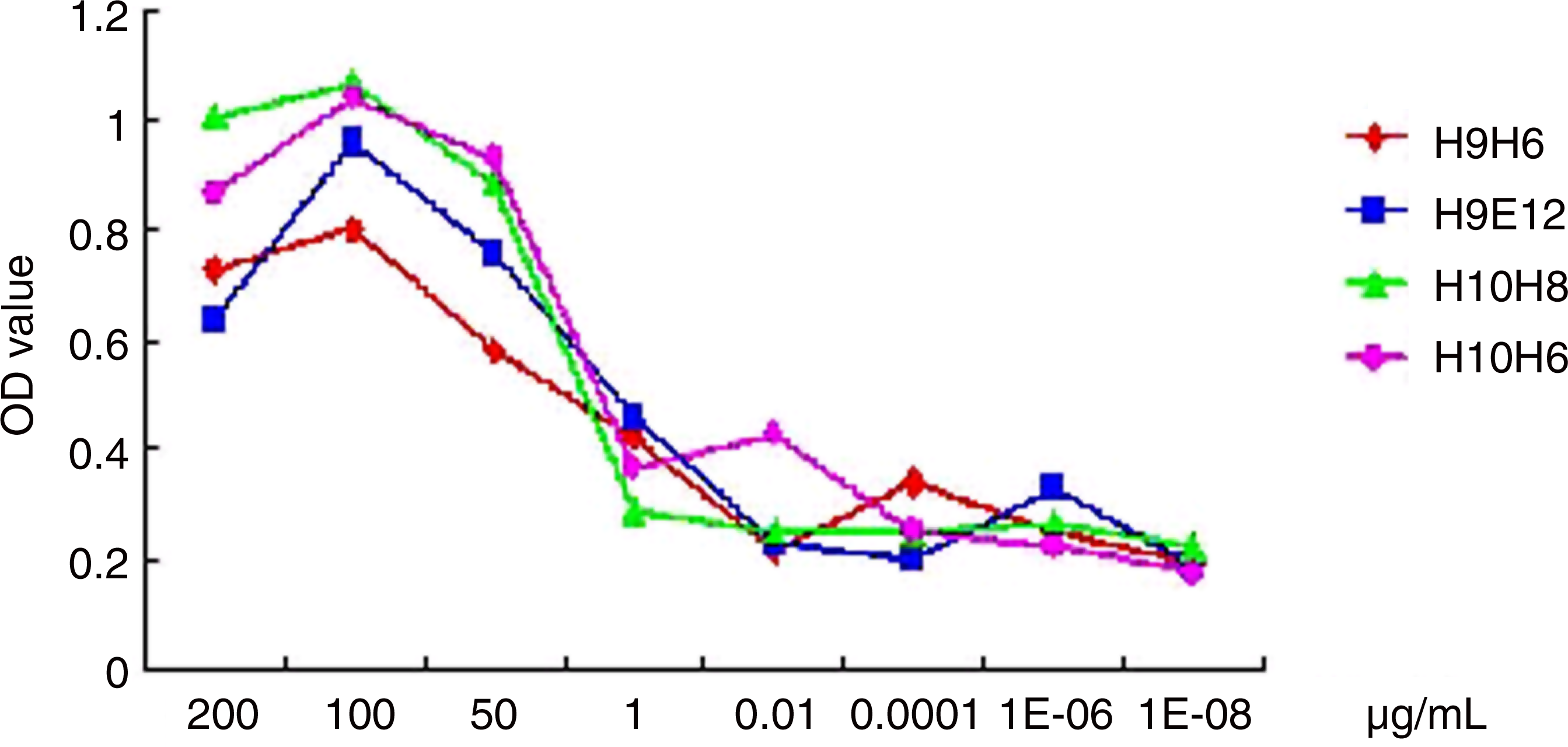
Relative affinity of the four MAbs determined by ELISA.
Epitopic topological relationships of MAbs against His6-FurA protein
In order to analyze whether the four MAbs recognize the same antigenic site, the AI values were detected by the ELISA additivity test. As shown in Table 2, all MAbs giving low self-AIs (ranging from 2.2% to 25.4%) confirmed that the antigen was efficiently saturated with all MAbs. The AI values calculated for pairs of MAbs suggested that the four MAbs shared the same antigenic epitopes of the His6-FurA protein.
The AI value was calculated according to the following formula: AI = [2 × A1+2/(A1 + A2) − 1] × 100%, where A1 + A2 are the ODs obtained when the MAbs are assayed separately, and A1+2 is the OD when the same amounts of the two MAbs are pooled in the same well in the additivity tests. For each pair of antibodies, the AI value is shown in percents. The AI value (<40%) indicates that the pair of antibodies recognize the same or adjacent epitope.
For each ascitic fluid, the concentration indicated in parentheses is sufficient to saturate the coated antigen.
Transcription analysis of furA gene in transfected COS-7 cells
It is known that antibodies that recognize proteins in their denatured form (SDS-PAGE gel) are not always able to detect the same proteins in a native form, and vice versa.(41) To determine if the prepared antibodies detect endogenic FurA in eukaryotic cells, the antibodies were used in lysate of Cos-7 cells transfected with pcDNA3.1(-)-furA. The results indicated that a protein of ∼20 kDa was detected by Western blot analysis using four MAbs (Fig. 5A). In contrast, no band was visualized in the Cos-7 cells transfected with pcDNA3.1(-) using either antibody. The results indicated that these four MAbs are specific to FurA. With the prepared MAbs, we detected the subcellular localization of FurA in the Cos-7 cells transfected with pcDNA3.1(-)-furA by immunofluorescence staining. Hoechst 33258 was used as a marker for the nuclei. The results of H9H6 (Fig. 5B) show that in the Cos-7 cells transfected with pcDNA3.1(-)-furA, FurA was localized in the cytoplasm along the cytoskeleton and in the nucleus. The same results were achieved using H9E12, H10H6, and H10H8 MAbs, which we prepared (data not shown). This indicated that all four MAbs could react specifically with the native FurA protein with good binding capacity.
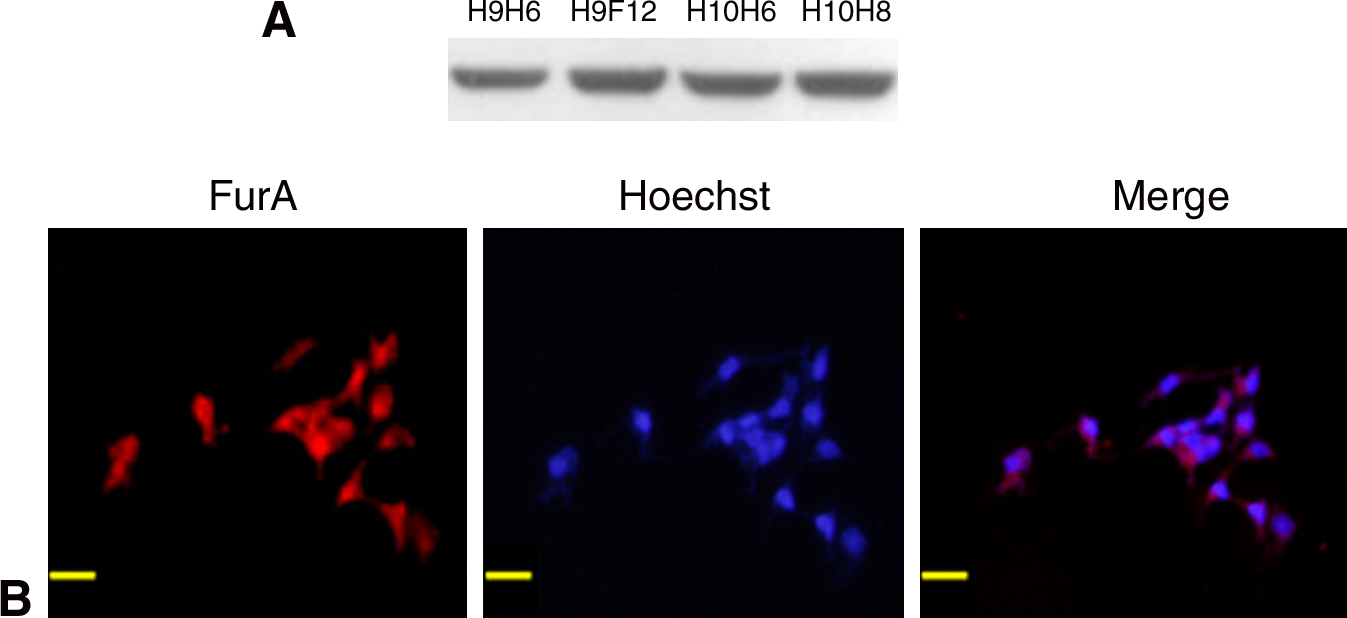
Expression and immunofluorescent co-localization of FurA in the transfected Cos-7 cells. (
Regulation of FurA by iron
Because members of the Fur family affect the expression of an array of genes involved with iron acquisition and storage, we reasoned that FurA had a role in regulating iron uptake in M. tuberculosis. Therefore, reducing or increasing the amount of iron available in the medium might lead to the difference of FurA protein expression. We assessed the effect of ferrous iron on the expression of FurA protein under three kinds of media (no iron, rich iron, and normal) by Western blot analysis (using H10H6 MAb). The results showed no significant difference when the strains were grown under the above conditions (Fig. 6).

Comparison of FurA expression in MTB H37Rv grown in r7H9, 7H9, and n7H9 media. Cell lysates of MTB H37Rv were incubated with H10H6 MAb.
Discussion
In an effort to obtain a good tool to elucidate the functional involvement of MTB FurA, four MAbs (H9E12, H10H6, H10H8 and H9H6) against MTB H37Rv FurA were prepared in the present study. In order to obtain abundant target protein to generate MAbs against MTB, prokaryotic expression was used. Recombinant proteins were highly and efficiently expressed in E. coli. The purified recombinant protein was used as immunogen to induce the production of MAbs. Western blot analysis and immunofluorescence results suggested that all four MAbs can recognize MTB FurA protein and serve as a good tool for its biological investigation.
Metalloproteins play structural and catalytic roles in gene expression.(42) The metalloregulatory proteins are a subclass that exerts metal-responsive control of genes involved in respiration, metabolism, and metal-specific homeostasis of stress-response systems, such as iron uptake and storage, copper efflux, and mercury detoxification.(43) The expression of a wide range of genes is controlled by metalloproteins. Metalloregulatory proteins are then a subset of regulatory proteins that act, at the physiological level, as components of metal-responsive genetic switches.(44) In the broadest sense, a metalloregulatory protein can serve a sensory or regulatory role in a switching mechanism. In some cases, a single protein plays both roles.(42)
Fur was initially discovered as a transcriptional repressor of a large number of genes for iron uptake system in response to iron sufficiency in E. coli. In the presence of divalent metal ions, such as ferrous iron, Fur proteins bind to a 19 bp imperfect dyad repeat (Fur-box) and inhibit transcription.(45) In addition to iron uptake systems, the Fur regulon includes genes for oxidative stress response. The furA-katG linkage was found to be conserved in most of the mycobacteria species. MTB FurA was 65% identical to M. smegmatis.(46) Ward and associates demonstrated that furA gene was induced in samples containing a high concentration of Cu. Cu-induced transcriptome have a strong correlation with genes induced by oxidative stress responses, especially when toxic levels of Cu were used.(47)
Although metal binding and activation mechanism have now been examined in greater detail for a significant number of Fur family members, including FurA and FurB, their exact mode of DNA-binding and in particular the structural basis of specific target recognition, remains unknown. Further knowledge about the architecture and function of Fur operator complexes will certainly enhance future structurally driven drug design efforts.
To investigate the exact function of FurA in the mechanism of MTB growth and pathogenicity, we amplified furA gene from the MTB H37Rv strain. In order to improve the efficiency of ligation with PCR products, furA full-length was first cloned in pGEM-T-Easy vector, which was supplied linearized with single 3′-T overhangs for TA cloning. These single 3′-T overhangs at the insertion site prevent recircularization of the vector and provide a compatible overhang for PCR.
BL21(DE3) was chosen as host cells to perform prokaryotic expression because it provides phage T7 RNA polymerase for expression of heterologous genes.(48) Using E. coli BL21(DE3) as host cells, we optimized the culture and induction parameters to get more soluble fusion proteins. We tried different IPTG concentrations (0.1, 0.2, 0.3, 0.4, 0.6, 0.8, 1.0 mM) and different temperatures (25, 30, 33, and 37°C) and found that an induction condition of 0.1 mM IPTG for 3 h at 33°C with a cell density OD600 reached 1.0 was the optimal system for the prokaryotic expression of MTB FurA.
The introduction of hybridoma technology has opened new vistas of research in immunology, diagnosis of infectious disease, and purification of specific antigens. A large number of MAbs have been raised to react with MTB antigens. MAbs to MTB have many applications: they can help us understand the pathogenicity of MTB, detect antigens and epitopes that may well contain species-specific and protective MTB determinants, provide specific skin test reagents, or define representative molecules which are possible targets for drugs. We continued and expanded research on MAbs against the MTB FurA protein to explore the potential use of the FurA protein in understanding the pathogenicity of MTB or search for possible targets of drugs and vaccines. We used purified FurA protein as immunogen to induce the production of MAbs and generated four MAbs.
All four MAbs reacted with the His6-FurA specifically, while they did not react with the irrelevant protein (His6-FurB). This suggests that MAbs recognize the epitopes on FurA, though not on His6. Titer assay illustrated that the MAbs generated by the H10H6 had the highest titers in both ascitic fluids and purified products. Epitopic topological relationships analysis suggested that the four MAbs shared the same antigenic epitopes of the His6-FurA protein. Furthermore, we assessed the effect of ferrous iron on the expression of FurA protein and concluded that in MTB H37Rv the expression of FurA protein is not influenced by the iron and it is not involved in iron uptake. Iron serves as a cofactor for FurA to regulate other gene expression. The biological role of FurA, in contrast to the role of most members of the family, appears to be more specialized.
In conclusion, a full-length furA DNA sequence with 525 bp from MTB H37Rv was cloned and demonstrated high expression in E. coli, and four highly specific and sensitive MAbs against FurA were produced. These results provide a substantial base for the further study of FurA in elucidating the potential role of the FurA protein in understanding the pathogenicity of MTB or research for possible targets of drugs and vaccines. All the results suggest that the four MAbs produced in this study have good bioactivity and reactivity with the FurA protein.
Footnotes
Acknowledgment
We thank Dr. Guangchun Bai of Wadsworth Center (New York State Department of Health) for his ongoing support and many valuable suggestions. This work was supported by the National Natural Science Foundation of China (no. 30470097).
Author Disclosure Statement
The authors have no financial interests to disclose.