
Abstract
HER2 proto-oncogene encodes a transmembrane receptor tyrosine kinase overexpressed in a variety of solid tumors. Several mouse monoclonal antibodies (MAbs) have been developed that recognize the extracellular part of HER2; of them two MAbs were humanized and employed for targeted immunotherapy. In this study we aimed to produce murine MAbs that specifically recognize the extracellular domain of human HER2. BALB/c mice were first primed with HER2-transfected NIH-3T3 cells and then boosted with recombinant extracellular part of HER2. Splenocytes from hyperimmunized mice were fused with myeloma cells and growing hybridomas were selected and screened for HER2 reactivity by an indirect ELISA. HER2-specific hybridomas were selected, cloned by limiting dilution assay, and further characterized by Western blotting and flow cytometry techniques. All clones showed positive reactivity to HER2 with binding affinity, ranging from 1.9 × 108 to 5 × 109, and stained HER2-transfected cells and malignant cells overexpressing HER2. None of the MAbs inhibited the binding of trastuzumab (Herceptin®) to HER2, indicating recognition of distinct epitopes by these MAbs. Based on these findings, our MAbs could be potentially used for selective targeting of HER2-expressing malignancies.
Introduction
Some monoclonal antibodies (MAbs) specific for the extracellular part of HER2 have been developed that specifically inhibit the growth of tumor cell lines overexpressing HER2. The well-known example is the murine 4D5 MAb, produced by Fendly and colleagues,(10) which was later humanized and designated as trastuzumab.(11) Trastuzumab or Herceptin® is the only anti-HER2 MAb approved by the Federal Drug Administration (Ministry of Health and Medical Education of Iran) for treatment of patients with HER2-overexpressing breast cancers.(12) Another MAb produced by the same group, 2C4, was found to inhibit the growth of breast cancer cell lines even with low expression of HER2.(9) This MAb was also humanized (pertuzumab),(13) and is currently being evaluated in a phase II clinical trial in combination with Herceptin as an adjunct to chemotherapy in patients with HER2-positive breast cancer.(14)
Here we report the development and characterization of novel mouse MAbs that specifically recognize the extracellular part of human HER2 molecule.
Materials and Methods
Cell culture
HER2-overexpressing human breast cancer cell line BT-474 was purchased from National Cell Bank of Iran (NCBI, Tehran, Iran) and cultured in RPMI 1640 culture medium (Gibco Invitrogen, Carlsbad, CA) containing 15% fetal bovine serum (FBS) (Gibco Invitrogen), 10 μg/mL insulin (Exir Co., Boroojerd, Iran), 100 μg/mL streptomycin, and 100 U/mL penicillin (Gibco Invitrogen), and incubated at 37°C in humidified atmosphere containing 5% CO2. NIH-3T3 and also HER2-expressing NIH-3T3 (provisionally named 3T3-HER2) were cultured under similar conditions, but in culture medium containing 10% FBS without insulin.
Stable transfection of HER2 gene
To develop a stable transfectant expressing HER2, human HER2 cDNA (OriGene, Rockville, MD) was amplified using Plasmid Maxiprep (Qiagen, Stockholm, Sweden). The construct was digested by 10 U NotI restriction enzyme (Invitrogen) and HER2 cDNA was ligated to NotI-digested pCMV6-Neo vector (OriGene). After selection of positive colonies with correct orientation, selected colonies were subjected to Maxiprep preparation. Transient expression of HER2 in NIH-3T3 cells was obtained by transfection of cells with 3 μg DNA in combination with 6 μL jetPEI™ DNA Transfection Reagent (Polyplus-transfection, New York, NY) according to the manufacturer's recommendations. After 48 h, cells were analyzed by Western blot analysis for HER2 expression. Transfected cells were subsequently selected using gradually increasing concentrations of G418 (Gibco Invitrogen) to 1500 μg/mL. At the end of selection, cells were analyzed by Western blotting and flow cytometry.
Immunization protocol and MAb production
We used both 3T3-HER2 cells and purified recombinant extracellular part of HER2 (Bender MedSystems, Vienna, Austria) to immunize mice. Six- to 8-week-old female BALB/c mice were immunized intraperitoneally with 5 × 106 3T3-HER2 cells in sterile PBS on weeks 0, 2, and 4.(10) Afterward they were injected subcutaneously with 2 μg recombinant extracellular part of HER2 in conjunction with complete Freund's adjuvant (CFA, Sigma, St. Louis, MO) and then with 1 μg in combination with incomplete Freund's adjuvant (IFA, Sigma) three times in 2-week intervals. The immunized mice were screened for serum HER2 antibody titer by indirect ELISA using recombinant extracellular HER2. Hyperimmunized mice were then injected intravenously with 2 μg recombinant extracellular HER2. After 3 days, splenocytes were harvested and fused with mouse myeloma cell line SP2/0 (National Cell Bank of Iran). Splenocytes were mixed at a 4:1 ratio with SP2/0 cells in the presence of polyethylene glycol (PEG) 1500 (Sigma). Fused cells were grown in hypoxanthine-aminopterin-thymidine (HAT selective medium, Sigma) and cloned by limiting dilution assay.
Supernatants of growing cells were screened first by indirect ELISA and all positive clones were further subcloned. Screening of subclones was done by sandwich ELISA and selected clones were tested again by indirect ELISA. Isotype of heavy and light chains of selected clones was determined using IsoStrip mouse monoclonal antibody isotyping kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's recommendations. Isotype of positive clones was confirmed by ELISA using subclass-specific antibodies (as described below). All clones were injected intraperitoneally to BALB/c mice 1 week after injection of 0.5 mL pristane (Sigma) to obtain ascitic fluid. Ascitic fluids were harvested, and MAbs purified using HiTrap™ SPG column (GE Healthcare, Freiburg, Germany).
Indirect enzyme-linked immunosorbent assay
All reactions were performed in sealed microtiter polystyrene plates (Maxisorp, Nunc, Roskilde, Denmark) in a volume of 50 μL. Plate washing was performed three times after each incubation with PBS (0.15 M [pH 7.2]) containing 0.05% Tween-20 (PBS-T, Sigma) for 1.5 h at 37°C. The plate was first coated with 0.5 μg/mL recombinant extracellular part of HER2 in PBS and incubated overnight at 4°C. After washing, plate was blocked using blocking buffer (PBS-Tween-20 containing 3% non-fat skim milk) at 37°C for 1.5 h. Then, supernatants of hybridomas were added at 37°C for 1.5 h. Herceptin (Genentech, South San Francisco, CA) was used as positive control. The washing step was repeated and horseradish peroxidase (HRP)-conjugated sheep anti-mouse Ig (HRP-conjugated sheep anti-human Ig for detecting Herceptin, prepared in our lab) was added and plate incubated for 1.5 h at 37°C. Following the final washing, the reaction was revealed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Sigma). Finally, the reaction was stopped by 20% H2SO4 and the optical density (OD) measured by a multiscan ELISA reader (Organon Teknika, Turnhout, Belgium) at 450 nm.
Sandwich ELISA
General considerations about the ELISA procedure have been given above. 2.5 μg/mL of Herceptin in PBS was adsorbed on wells of a micotiter ELISA plate overnight at 4°C. After the washing step, 1:15 dilution of BT-474 cell lysate in blocking buffer was added and incubated for 1.5 h at 37°C. The washing step was repeated and supernatants of hybridomas were added and plate was incubated for 1.5 h at 37°C. The plate was washed and HRP-conjugated sheep anti-mouse Ig (human Ig-adsorbed, prepared in our lab) was added and incubated for 1.5 h at 37°C. After washing, TMB substrate was added, the reaction was stopped by H2SO4, and ODs were measured as mentioned above.
Isotyping of selected MAbs by ELISA method
0.5 μg/mL of each goat anti-mouse IgG1 and IgG2a isotypes (Sigma) were coated separately in PBS overnight at 4°C in ELISA microtiter plate. The plate was washed and after blocking for 1.5 h at 37°C and washing, supernatants of selected clones were added and incubated for 1.5 h at 37°C. Washing step was done again, and all wells were incubated with HRP-conjugated sheep anti-mouse Ig for 1.5 h at 37°C. Following the addition of TMB substrate solution and then stopping solution, the ODs were measured.
Western blot analysis
BT-474, NIH-3T3, and 3T3-HER2 cells were harvested and total cell lysates were prepared by adding 1 mL/107 cell lysis buffer (10 mM Tris [pH 7.4], 100 mM NaCl, 1 mM EDTA, 1 mM NaF, 20 mM Na4P2O7, 1% Triton X-100, 10% glycerol, 0.1% SDS) containing 10% protease inhibitor cocktail (Sigma). After 1 h incubation on ice, cell lysates were centrifuged at 10,000 g for 10 min. Supernatants were collected and total protein concentrations were measured by BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, IL) according to the manufacturer's recommendations. 40 μg of total cell lysates was separated on 10% SDS-PAGE and electroblotted on PVDF membrane (Roche Diagnostics, Mannheim, Germany). After blocking of membrane with blocking buffer (PBS-Tween-20 containing 5% non-fat skim milk) overnight at 4°C and washing three times with washing buffer (0.05% Tween-20, 1 × PBS) for 15 min, primary antibodies were added at 10 μg/mL in blocking buffer at room temperature for 1.5 h while shaking. 10 μg/mL of each Herceptin and anti-HIV Env protein MAb with IgG1 isotype (produced in our lab) were used as positive and negative controls, respectively. Washing steps were repeated and HRP-conjugated sheep anti-mouse immunoglobulin (HRP-conjugated sheep anti-human Ig for detecting Herceptin, prepared in our lab) was added at room temperature for 1.5 h on shaker. After washing, bands were visualized using 3,3′-diaminobenzidine tetrahydrochloride (DAB) substrate (Sigma).
Flow cytometry
The indirect staining was performed on BT-474, 3T3-HER2, and NIH-3T3 cells at surface level. After harvesting the cells by trypsinization and twice washing with washing buffer (PBS, 0.1% NaN3), 106 cells were incubated with 100 μL of 10 μg/mL of SPG-purified anti-HER2 antibodies as primary antibodies at 4°C for 1 h. SPG-purified mouse anti-HIV Env MAb with IgG1 isotype (produced in our lab) was included as negative control. After incubation, the cells were washed twice with washing buffer and then incubated with FITC-conjugated sheep anti-mouse immunoglobulin (prepared in our lab) at 4°C for 1 h. Cells were then washed twice before scanning with a flow cytometer (Partec, Nuremberg, Germany). Data analysis was performed using Flomax flow cytometry analysis software (Partec).
Affinity constant determination
An ELISA-based method was employed to determine affinity constant of our MAbs.(15) Briefly, 1, 0.5, 0.25, 0.625, 0.312, and 0.156 μg/mL recombinant extracellular part of HER2 was coated on wells of a micotiter ELISA plate. Blocking step was performed as described earlier for the indirect ELISA method. Serial concentrations of MAbs (1 − 0.015 μg/mL) in blocking buffer were added into each coated well and incubated at 37°C for 1.5 h. After the washing step, all wells were incubated with HRP-conjugated sheep anti-mouse Ig for 1.5 h at 37°C. Washing steps were repeated and ODs were measured following addition of TMB substrate solution and then stopping solution. Sigmoidal curves of ODs versus the logarithm of antibody concentrations were drawn. The antibody concentration resulting in 50% of the maximum absorbance value ([Ab]t) at a particular antigen coating concentration was selected for the affinity calculation using the formula Kaff = 1/2(2 [Ab´]t - [Ab]t). [Ab´]t and [Ab]t represent the antibody concentrations resulting in 50% of the maximum absorbance value at two consecutive concentrations of coated antigen where [Ag] = 2 [Ag´]. Final Kaff value was the mean of such calculations for three antigen concentrations.
Assessment of epitope specificity of MAbs by competition ELISA
ELISA plate was coated with 0.5 μg/mL recombinant extracellular part of HER2 and then blocked as previously described. All MAbs in final concentrations of 50, 12.5, 3.1, 0.8, 0.2, 0.5, and 0.012 μg/mL were added together with 0.5 μg/mL of HRP-conjugated Herceptin (prepared in our lab) and incubated at 37°C for 1.5 h. Washing steps were repeated and ODs were measured after adding TMB substrate solution and then stopping solution. Serial dilutions of unconjugated Herceptin alone were used as positive control for this assay. Percent of inhibition for each MAb was calculated based on the following formula: percent of inhibition = [(ODNI − ODWI)/ODNI] × 100, where ODNI represents OD obtained for Herceptin binding without competitor MAb and ODWI represents OD obtained for Herceptin binding in presence of competitor MAb.
Results
Establishment of stable HER2-expressing NIH-3T3 cells
HER2-negative mouse NIH-3T3 cell line was transfected by HER2-pCMV6-Neo construct using jetPEI transfection reagent. After 48 h, transient expression of human HER2 protein was assessed by Western blotting. Cell lysates of HER2-overexpressing BT-474 cell line and mock-transfected NIH-3T3 cells were employed as positive and negative controls, respectively (Fig. 1A). Stable transfectant (3T3-HER2) was selected by G418 and shown to be HER2 positive by flow cytometry (Fig. 1B).
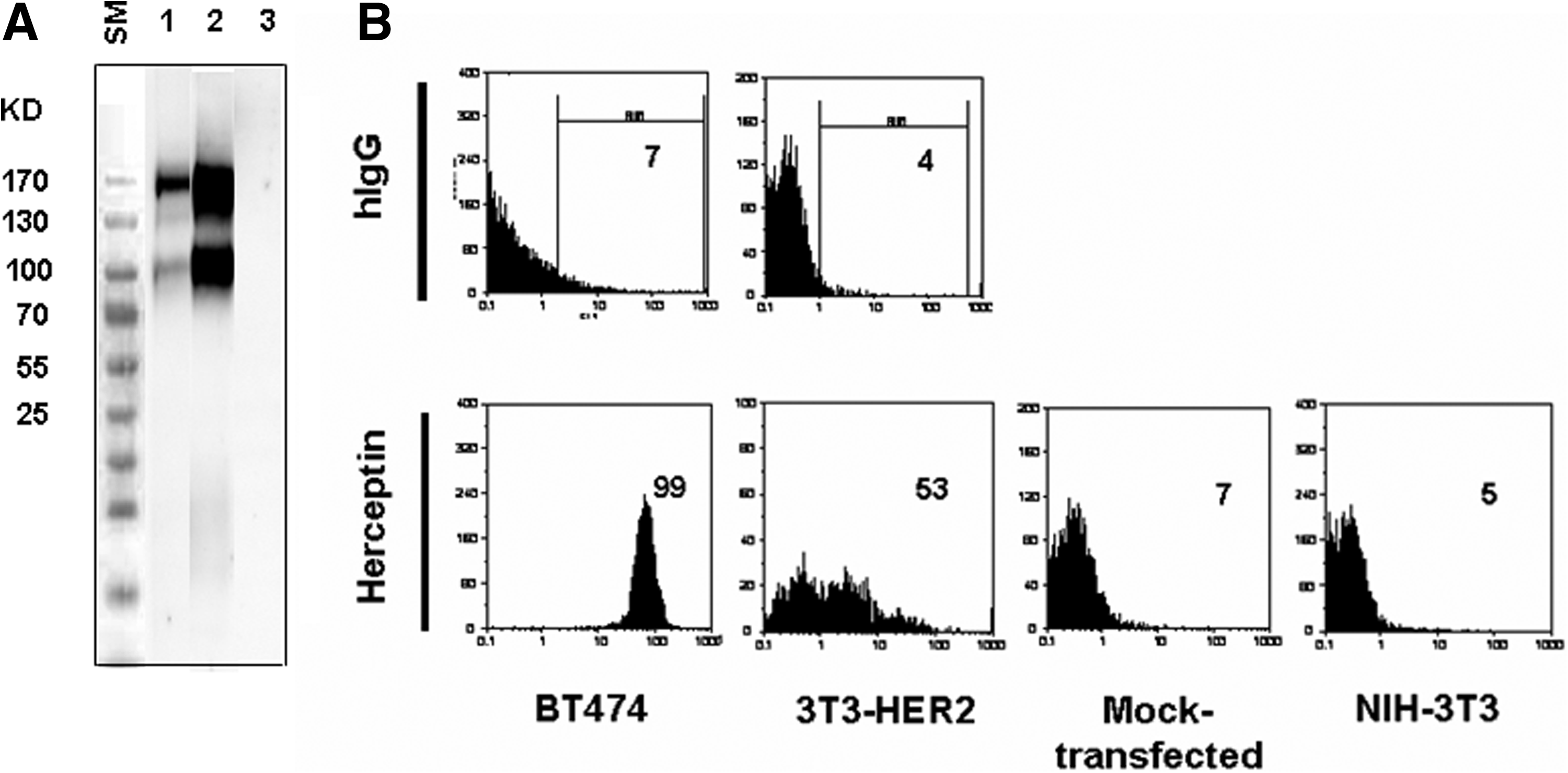
HER2 expression in transiently transfected NIH-3T3 cells. (
Production of anti-HER2 MAbs
After completion of immunization of mice with 3T3-HER2 cells followed by recombinant extracellular HER2 protein, high titer of anti-HER2 antibody in serum of immunized mice was detected by ELISA (Fig. 2). Splenocytes of hyperimmunized mice were fused with SP2/0 cells and all growing hybridomas were initially screened based on reactivity to recombinant extracellular part of HER2 by an indirect ELISA. By this screening assay, all non-reactive clones, including those recognizing intracellular part of HER2, were excluded. Five hybridomas were finally found to react with recombinant extracellular HER2 by indirect ELISA, including 1F2, 2A8, 1H6, 4C7, and 2A9. Isotype of MAbs secreted by all clones was determined by IsoStrip isotyping kit. Three of the clones (2A8, 1H6, and 4C7) were found to be IgG1κ, whereas the other 2 clones (1F2 and 2A9) displayed IgG2a κ isotype. IgG1 and IgG2a subclasses of the MAbs were also confirmed by ELISA using mouse subclass-specific polyclonal antibodies (Table 1).
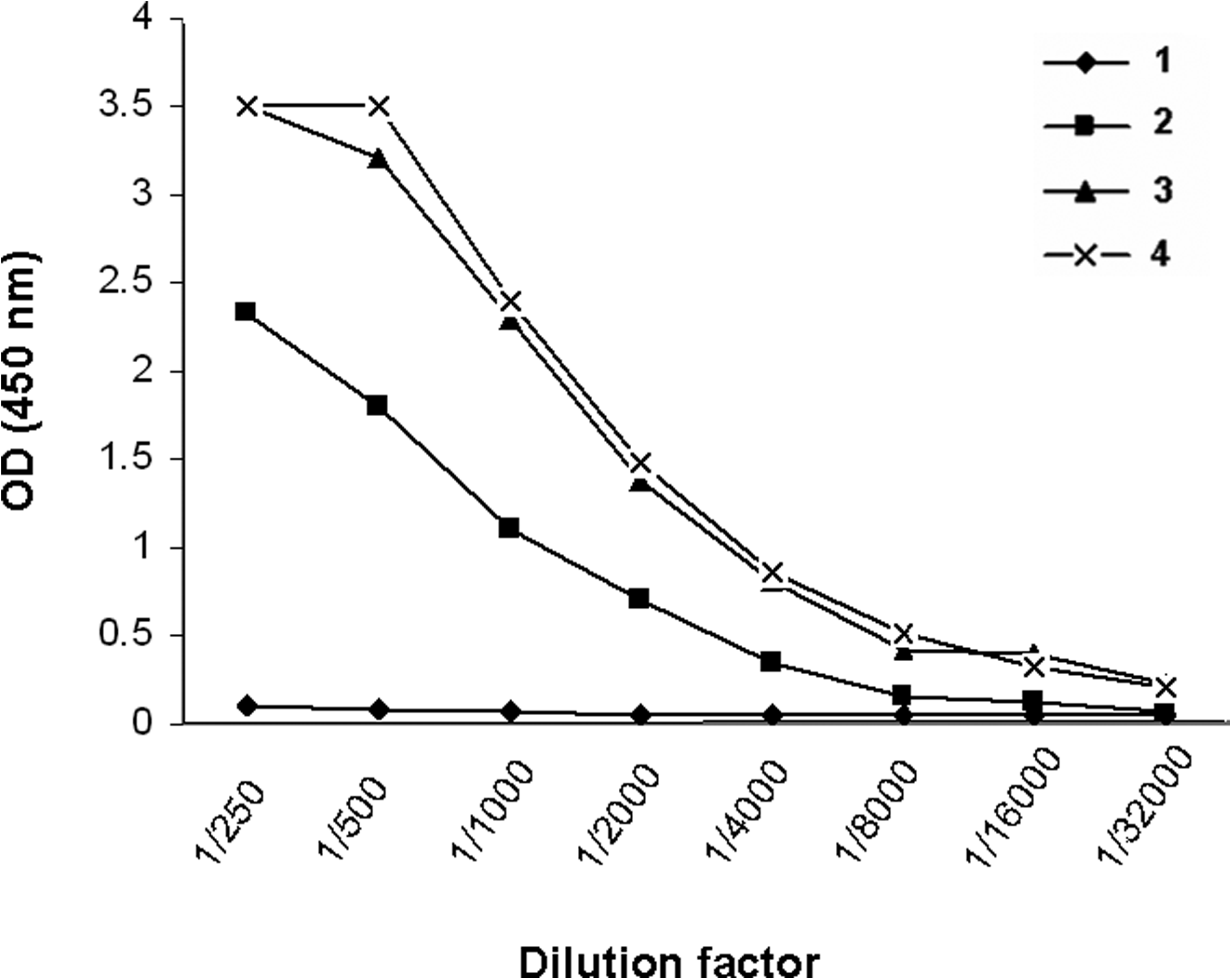
Titration of specific anti-HER2 antibody in serum of immunized mice. Serum samples collected at different time intervals after onset of immunization were serially diluted on recombinant extracellular HER2 coated plates. (1) Preimmune serum, (2) 2 weeks after the last immunization with 3T3-HER2 cells, (3) 2 weeks after the first immunization with recombinant extracellular domain of HER2, and (4) 2 weeks after the last immunization by recombinant extracellular domain of HER2.
The results represent OD values read at 450 nm. Cell culture medium was used as negative control. Clones 1H9, 1T0, and 1B5 represent HER2-specific MAbs with known isotype already prepared in our laboratory.
Affinity constant (Kaff) for each selected concentration of antigen and antibody was determined using the formula described in Materials and Methods.
Specific reactivity of MAbs to HER2 molecule
For further characterization of anti-HER2 MAbs, Western blot and flow cytometric analyses were performed. In parallel to trastuzumab (Herceptin) as positive control, all MAbs showed positive reactivity and detected two 185 and 95 − 100 kDa bands (Fig. 3), corresponding to full-length and truncated forms of the HER2 receptor, respectively.(16) Additionally, all MAbs showed positive surface staining with BT-474 and 3T3-HER2, but not NIH-3T3 cell lines (Fig. 4). The affinity constant (Kaff) of all MAbs was determined by an ELISA-based assay. The range of Kaff varied between 1.9 × 108 to 5 × 109 (Table 1).
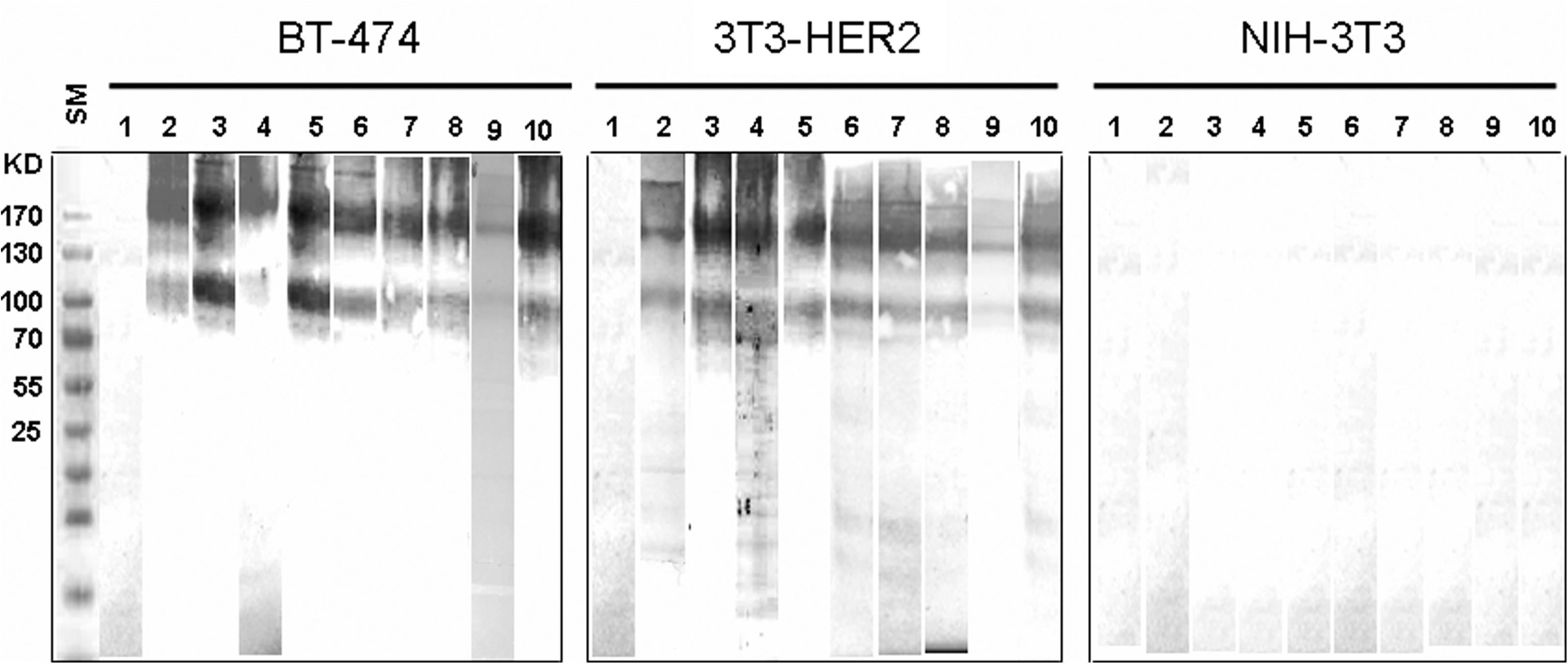
Reactivity of selected MAbs to human HER2 molecule by Western blot analysis. Cell lysates of BT-474, 3T3-HER2, and NIH-3T3 cell lines were separated on 10% SDS-PAGE gel electrophoresis. After electroblotting to PVDF membrane, all samples were probed by SPG-purified anti-HER2 MAbs. Development of color by DAB chromogen showed that all MAbs recognize human HER2 as two 185 and 95 − 100 kDa bands in BT-474 and 3T3-HER2 cell lysates, but not in NIH-3T3. Lanes represent: (1) anti-HIV Env as negative control, (2) Herceptin as positive control, (3) 1B5, (4) 1T0, (5) 1F2, (6) 2A8, (7) 1H6, (8) 4C7, (9) 2A9, and (10) 1H9. SM, protein size marker.
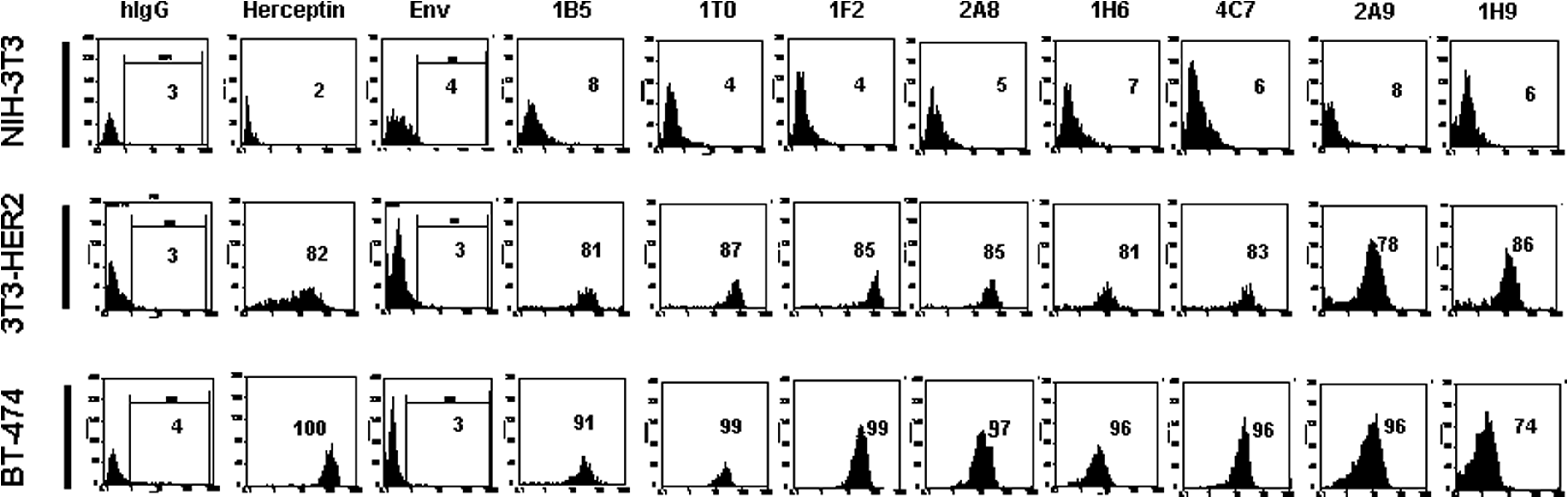
Detection of surface expression of human HER2 by flow cytometry. Values presented in figure represent percent of positive cells. Adherent HER2-expressing BT-474 and 3T3-HER2, and also NIH-3T3 cells were harvested and stained with SPG-purified MAbs. SPG-purified mouse anti-HIV Env MAb with the same concentration was used as negative control. All clones detected transmembrane human HER2 on the surface of BT-474 and 3T3-HER2 cells, but not on NIH-3T3. Herceptin and SPG-purified human IgG (hIgG) were used as positive and negative controls.
Competition of MAbs with Herceptin for HER2 binding
HER2 binding inhibition of Herceptin by our MAbs was investigated by a competition ELISA. While as low as 1 μg/mL of unconjugated Herceptin induced approximately 50% inhibition, none of the MAbs employed at the highest concentration (50 μg/mL) revealed this level of inhibition. Only 2A8 and 1H9 were able to show 20% inhibition when used at this concentration. Representative results obtained for 2A8, 1H9, and 4C7 are depicted in Figure 5. Unconjugated Herceptin was also included as a control to obtain maximum inhibition.
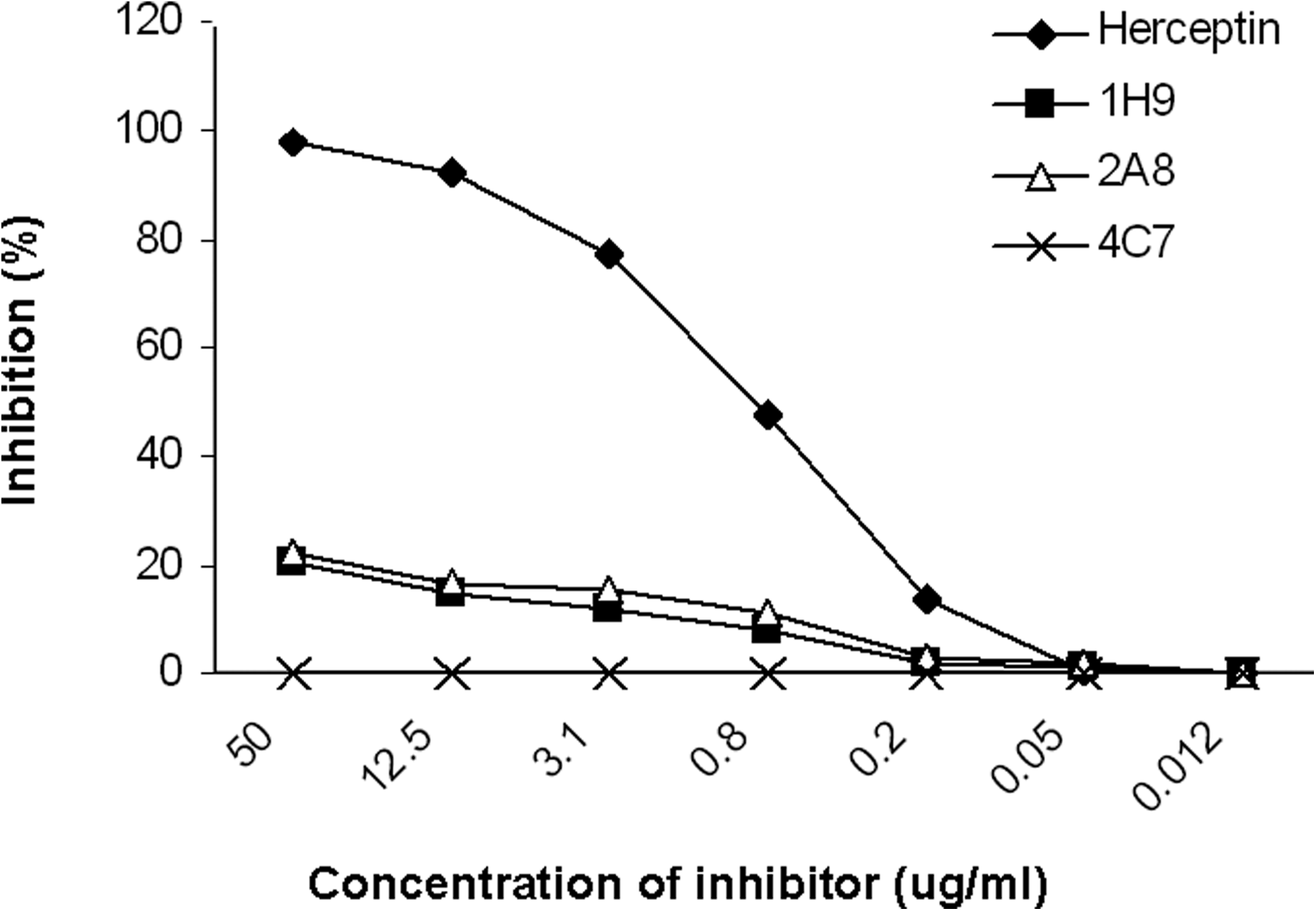
Representative results for cross-inhibition of Herceptin by anti-HER2 MAbs HRP-conjugated Herceptin and different concentrations of anti-HER2 MAbs were allowed to compete for binding to constant concentration of recombinant extracellular domain of HER2 in 96-well ELISA plate. Unconjugated Herceptin was used as positive control for competition assay.
Discussion
Several studies have shown overexpression of HER2 in a number of human malignancies.(4–7) Owing to tyrosine kinase activity of this transmembrane receptor,(1) HER2 molecule has been considered a potential target for specific immunotherapy of HER2 positive malignancies.(17) Several approaches have been proposed for treatment of such malignancies, including induction of passive and active immunity against HER2 receptor by administration of MAbs recognizing the extracellular domain of the receptor(18) and active vaccination with HER2 protein, peptide and DNA.(19,20) To inhibit signaling of HER2 receptor, tyrosine kinase inhibitors (TKIs) have also been employed as single agents or in combination therapy.(21)
Monoclonal antibodies specifically directed against the extracellular domain of HER2 have long been considered for targeted immunotherapy.(22) Some investigators have developed anti-HER2 extracellular domain antibodies.(10,23–25) Ten HER2-recognizing MAbs have been developed, two of which were introduced as potential agents for targeted immunotherapy by Fendly and associates.(10) Of these, murine 4D5 and 2C4 MAbs were shown to inhibit growth of breast cancer cell lines with high and low expression of HER2, respectively.(9,26) Murine 4D5 was humanized and called trastuzumab or Herceptin.(11) Trastuzumab inhibits the growth of HER2-overexpressing malignant cells by downregulation of the receptor, prevention of heterodimer formation,(27) initiation of Gl arrest,(28) prevention of HER2 ectodomain cleavage,(29) inhibition of angiogenesis,(30) decreasing activation of the PI3K/Akt pathway in HER2 signaling process,(31) and triggering immune responses such as antibody dependent cell-mediated cytotoxicity (ADCC)(11) and complement-dependent cytolysis (CDC).(32) Humanized 2C4, called pertuzumab (Omnitarg),(13) inhibits ligand-dependent HER2 signaling and formation of HER2/HER3 complexes, even in low HER2-expressing human breast cancer cells.(9)
In the present study we reporte the development of new MAbs specifically recognizing the extracellular part of HER2 molecule by immunizing mice with both HER2-transfected NIH-3T3 cells and recombinant extracellular part of human HER2. All clones were reactive in sandwich and indirect ELISAs, Western blotting as well as flow cytometry. Our novel immunization protocol adapted in this study favors efficient immunization and development of MAbs recognizing the extracellular part of HER2. Initial immunization of BALB/c mice with HER2-transfected NIH-3T3 cells originated from the same strain of mouse is expected to elicit moderate antibody response to all epitopes located in the extracellular and cytoplasmic domains of the HER2 molecule without induction of antibody response to the cellular antigenic constituents. Boosting of mice with recombinant extracellular part of human HER2 allows hyperimmunization against the HER2 extracellular domain (Fig. 1). Additionally, initial screening of all growing hybridomas by indirect ELISA with recombinant extracellular part of human HER2 will select only extracellular domain, but not cytoplasmic domain-specific hybridomas. Thus, all clones reactive with the cytoplasmic epitopes of HER2 will be automatically excluded. A similar immunization schedule has been reported previously(10) using HER2-transfected NIH-3T3 and partially purified full-length HER2 from these cells. Partially purified HER2 was also used for screening of hybridomas. This strategy, however, is expected to induce weaker immunity and to select for more cross-reactive MAbs. Indeed, two of the HER2-specific MAbs failed to stain HER2 overexpressing SKBR3 cell line by flow cytometry.(10) Two other groups(23,24) reported production of only one murine MAb recognizing the extracellular domain of HER2 by immunizing mice with only HER2-transfected NIH-3T3. Kametani and colleagues(25) immunized non-obese diabetic/severe combined immunodeficient/IL-2 receptor γ null mouse (NOG) mice with HER2 extracellular domain and HER2-derived peptide and could obtain only one IgM-producing human hybridoma.
In an ELISA-based method, none of our MAbs showed substantial inhibitory potential to hinder Herceptin binding to extracellular part of human HER2 molecule. Exceptions were 2A8 and 1H9, inhibiting up to 20% at the highest concentration tested (50 μg/mL). Our results suggest that all our MAbs, even 2A8 and 1H9, which showed marginal inhibition, recognize distinct epitopes located not in proximity of the epitope recognized by Herceptin. This, together with the high binding affinity of some of our MAbs, suggests the potential implication of these MAbs for targeting of HER2 positive malignant cells. Further in vitro and in vivo analyses are required to assess the functional activity of our MAbs.
Footnotes
Acknowledgments
We would like to thank Simin Nobakht and Firouz Biglari for their technical assistance. This study was supported in part by grants from the Food and Drug Administration of the Ministry of Health and Medical Education of Iran, and the High Technology Schemes of the Ministry of Industries and Mines of Iran.
Author Disclosure Statement
The authors have no financial interests to disclose.