
Abstract
The chromosomal translocation t(8;21) often found in acute myeloid leukemia generates an oncogenic fusion protein AML1-ETO. This chimeric oncoprotein disrupts wild-type AML1 function and dysregulates genes important for normal myelopoiesis. Monoclonal antibodies that can capture and detect the AML1-ETO fusion protein would help with early diagnosis and treatment prognosis of acute myeloid leukemia. We report the development of murine monoclonal antibodies (MAbs) that specifically bind epitopes encoded by either AML1 or ETO. Since alignment to the human ETO protein indicated almost 100% homology to the mouse ortholog, a strategy was needed to instruct humoral immunity in mice to focus and respond to self-epitopes. Our strategy to develop capture/detector reagents involved producing MAbs that would bind to epitopes within the non-fused myelopic protein (i.e., either AML1 or ETO). This included a process to select antibodies for their ability to also recognize the translocated chromosomal AML1-ETO fusion protein and to identify complementary capture/detector antibody pairs. Construction of a peptide hapten-carrier complex and use of a rapid immunization protocol resulted in IgM-IgG ETO specific MAbs. These MAbs bound specifically to a recombinant form of AML1-ETO fusion protein expressed in HEK and to an endogenous AML1-ETO form of the fusion protein expressed in Kasumi-1. We report the development of murine hybridoma MAbs derived from immunizations with a peptide “self-epitope.” Our findings provide a potential strategy to instruct humoral immunity in mice to focus and respond to self-epitopes. This strategy has been validated with the oncogenic fusion protein AML1-ETO involved in acute myeloid leukemia.
Introduction
To generate high affinity MAbs to ETO, a protein that shares 99% amino acid homology between murine and human, the state of self-tolerance,(7,8) would have to be overcome. Several publications have described situations where self-tolerance was broken and either polyclonal(9–11) or monoclonal(12) immune responses to self-antigen occurred. In cancer, it is known that patients develop autoantibodies to cancer antigens(13–16) secreted from their tumors. It is also well established that antibody-mediated pathology goes unregulated in autoimmune diseases such as lupus and rheumatoid arthritis.(17) These examples led us to believe that it would be possible to break self-tolerance and develop murine hybridoma anti-self MAbs.
Productive humoral responses require both T-cell and B-cell collaboration.(18) Historically, B-cell haptens have been conjugated to large carrier proteins, such as keyhole lympet hemocyanin, to provide T-cell epitopes necessary for hapten-specific responses. More recently, polyclonal immune responses have been demonstrated after immunizations using a chimeric linear peptide containing a foreign T-cell epitope fused to a B-cell epitope.(19–21) Cloning of the T-cell epitope into a recombinant self-antigen also resulted in a polyclonal immune response.(11) We therefore designed a peptide with a foreign T-cell epitope sequence from the sperm whale myoglobin gene fused to a 100% conserved B-cell epitope from ETO. The sperm whale myoglobin peptide stimulates T-cells by fitting within the binding grove of MHC class II molecules on antigen-presenting cells of Balb/c (H-2d haplotype) mice.(22)
To maximize the T-cell and B-cell response to the ETO peptide, several adaptations were incorporated into our MAb development process. First, intrasplenic immunizations were performed to traffic immunoreactive lymphocytes into the spleen by potentially activating the CXCR5-CXCL13 chemokine pathway.(23) The CXCR5-CXCL13 chemokine pathway promotes and defines secondary lymphoid tissue follicular trafficking of naïve B-cells and activated T-cells.(24–26) Follicular helper T-cell localization in germinal centers is critical for high affinity B-cell maturation and differentiation into plasma and memory B-cells.(26) Second, the lack of myoglobin peptide-reactive T-cells that constitutively express CD154 could limit ETO-peptide-specific B-cell expansion. Exogenous anti-CD40 antibodies were used(27) to augment the CD40-CD154 cell signaling pathway response to a self-epitope. The CD40-CD154 signaling pathway is critical for humoral immune responses to thymus-dependent antigens.(28) Expansion of follicular B-cell populations by exogenous CD40 agonist antibody(27) would be expected to circumvent defects in follicular T helper cell CD154 expression during response to self-antigen as this treatment has restored full humoral response to adenovirus in CD154-deficient mice.(29)
The combination of intrasplenic immunization to traffic immunoreactive lymphocytes, presumably mediated by CXCR5-CXCL13, and expansion of follicular B-cell populations by exogenous CD40 antibody all provides a basis to focus the humoral immune response in the spleen. This report describes a novel process to generate an immune response to self-antigens and is exemplified by generation of anti-human ETO-specific MAbs from mouse hybridoma cell lines.
Materials and Methods
Construction of expression vectors
The AML1-ETO, ETO, and AML1 genes were amplified by polymerase chain reaction (PCR) using an oligo(dT)-primed cDNA preparation made from the Kasumi-1 cell line (DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany). The amplicons for AML1-ETO (sense primer 5′-TAG ACT GCG GCC GCC ACC
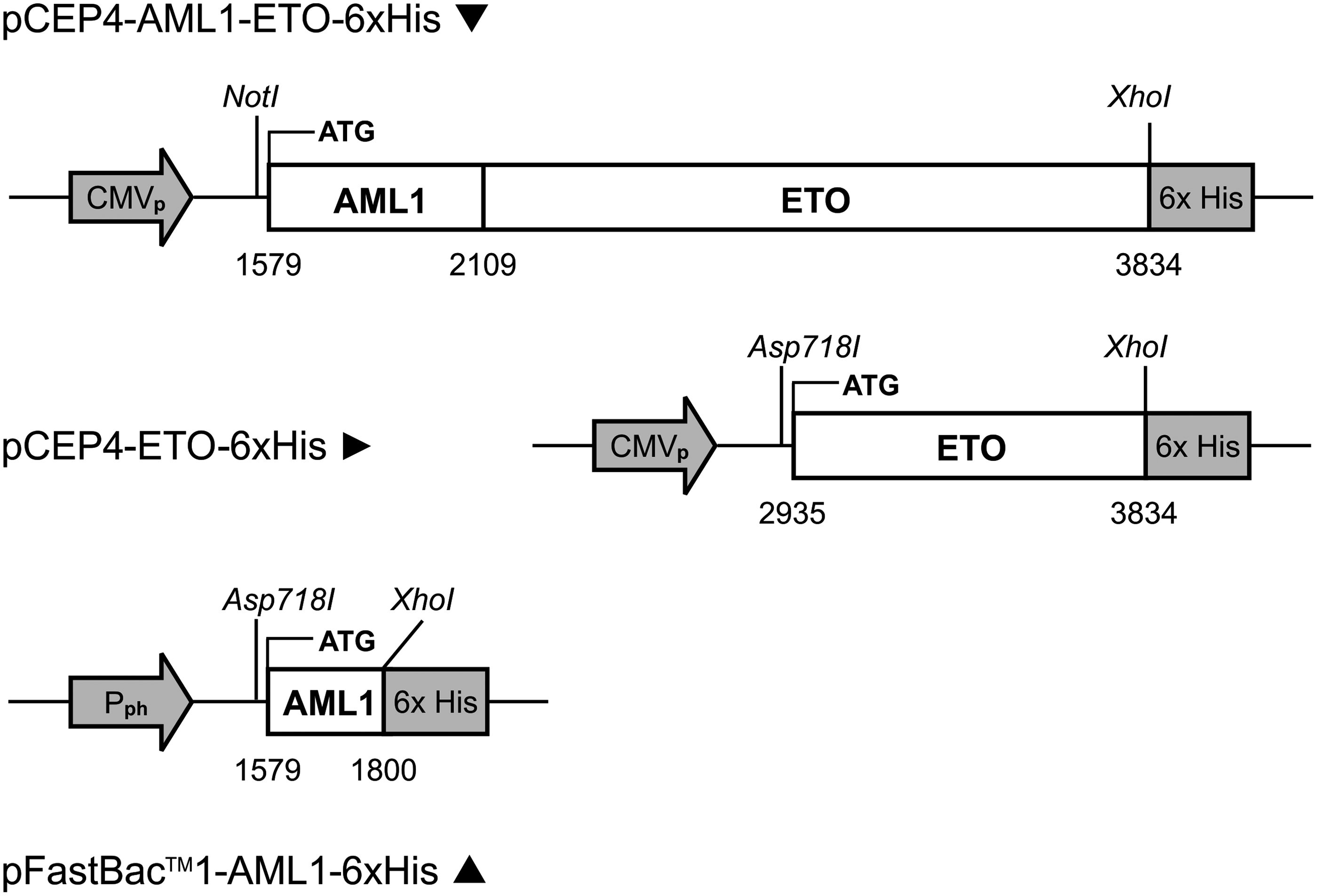
Schematic for AML1-ETO, ETO, and AML1 genes inserted into expression plasmids (pCEP4) or transfer vector (pFastBac™1). Numbering is based on nucleotide sequence for GenBank accession D13979. CMVp, cytomegalovirus promoter; Pph, polyhedrin promoter; 6xHis, coding sequence for hexahistidine epitope tag.
Expression of recombinant proteins in HEK 293 and insect cells
HEK cells (FreeStyle 293-F cells, Invitrogen) were grown as suspension cultures in serum-free medium (GIBCO FreeStyle™ Expression Media, Invitrogen). For recombinant protein expression, a mixture of 0.1 L of DMEM (cat. no. SH30319.01, Hyclone, Logan, UT), which contained complexes made by mixing together 0.5 mg of plasmid DNA and 1.5 mg of PEI (polyethylenimine, Sigma-Aldrich, St. Louis, MO), was used to transfect 0.9 L of cells at 1.1 × 106 cells/mL. At 3 days post-transfection, the cells were collected by low speed centrifugation (200 g). For recombinant AML1 protein expression, the Bac-to-Bac Baculovirus expression system was used according to the manufacturer's protocol.
Mice
Female SJL mice (Taconic Hudson, Germantown, NY) or Balb/c mice (Charles River Laboratories, Raleigh, NC) between 6 to 8 weeks old were obtained for use in antibody development. Mice were housed and immunized according to an approved institutional animal care and use committee protocol.
Chimeric ETO peptide construction
The sperm whale myoglobin amino acid sequence from 106–118 (FISEAIIHVLHSR) was used as a foreign T-cell epitope. The B-cell epitope DSSESCWNCGRKASE was derived from the C-terminus of human ETO. Single peptides were synthesized by New England Peptide (Gardner, MA) with the generic format: H2N-FISEAIIHVLHSR DSSESCWNCGRKASE–OH and provided as a lyophilized product. HPLC purity was >85%. Peptides were hydrated in DEPC water (Invitrogen) at 2.5 mg/mL and stored at 4°C.
Immunization of mice
Two different immunization strategies were used: repetitive immunization multiple sites (RIMMS) 30 and rapid intrasplenic immunization (IS).
SJL mice at 12 weeks of age were used for the RIMMS protocol. Prior to immunization, mice were anesthetized with 2% isoflurane (Butler Animal Health Supply, Dublin, OH) in an EZ anesthesia vaporizer (Palmer, PA) according to the manufacturer's instructions. Fifty micrograms of recombinant ETO B.003 were emulsified in either Freund's complete adjuvant (Sigma-Aldrich) and 5 μL Gerbu adjuvant MM (Gerbu Biotechnik, Gaiberg, Germany) for the initial day 0 immunization, or Titermax adjuvant (Norcross, GA) and 5 μL Gerbu adjuvant MM for day 4 and 11 immunizations. Multiple injections were made in the subcutaneous tissue (50 μL/site) to focus drainage into the popliteal, inguinal, axillary, and brachial lymph nodes. Mice were sacrificed on day 13, and the eight bilateral lymph nodes along with two lumbar lymph nodes were collected and pooled for fusion.
A Balb/c mouse at 12 weeks of age was used for rapid IS immunization. The mouse was anesthetized with 1.2 mg ketamine-HCL (Bioniche Animal Health, Athens, GA) and 0.39 mg xylazine (Lloyd Laboratories, Shenandoah, IA) injected intraperitoneally prior to immunization. The mouse was shaved with clippers to visualize the spleen under the skin on day 0. Thirty minutes after initial anesthesia the mouse was placed in 2% isoflurane gas until immobilized. One hundred micrograms of ETO chimeric peptide mixed with 5 μL Gerbu adjuvant were injected directly into the spleen in two sites in a total volume of 150 μL using a BD Ultrafine II insulin syringe (Franklin Lakes, NJ) on days 0, 4, and 11. Fifty micrograms of anti-CD40 agonist MAb clone 1C10 (R&D Systems, Minneapolis, MN) were injected in the subcutaneous tissue at the base of the tail in a 25 μL volume on day 10.(27) The mouse was sacrificed on day 13 and the spleen was collected for fusion.
Cell fusion
Lymphocytes were flushed from organs with 10 mL RPMI (Invitrogen), pelleted by centrifugation, and fused 1:1 with a stable Bcl-2 expressing P3X63.Ag8.653 myeloma cell line with 100 μL polyethylene glycol 1500 (Roche Diagnostics, Indianapolis, IN) per 107 lymphocytes. Cells were resuspended in selection medium (50% Ex-Cell™ 610-HSF [Lenexa, KS], 38% RPMI, 10% FBS [Hyclone], 1% penicillin-streptomycin-L glutamine, 1% non-essential amino acids [Invitrogen], 5.7 μM azaserine, 100 μM hypoxanthine [Sigma-Aldrich], 1 μg/L human IL-6) and seeded into 96-well tissue culture plates (Corning, Corning, NY) at 107 lymphocytes/plate for RIMMS or 2 × 106 lymphocytes/plate for IS fusions. Non-hybridoma immunoglobulin, derived from non-fused B-cells, was minimized by media replacement on days 7–9 post-fusion with feeding media (50% Ex-Cell 610-HFS, 33% RPMI, 1% penicillin-streptomycin-L glutamine, 1% non-essential amino acids, and 15% FBS). Resulting hybridomas were screened for target specificity by ELISA on day 12 using the immunogen and an irrelevant 6x histidine-tagged protein control. Positive cultures were expanded to 12-well tissue culture plates (Corning), grown to confluency in expansion media (50% Ex-Cell 610-HSF, 33% RPMI, 10% FBS, 1% penicillin-streptomycin-L glutamine, 1% non-essential amino acids, 1 μg/L human IL-6, 5% Hybridoma Cloning Factor [Bioveris, Gaithersburg, MD]), and frozen. Antibody containing supernatants were characterized by ELISA and Western blot analysis.
Anti-AML1 clone 7G3.7, used as a capture antibody in an AML1-ETO sandwich ELISA, was derived from previous work using the RIMMS strategy with recombinant AML-1. This clone was determined to be the best AML1-ETO capture antibody (data not shown).
Hybridoma characterization: ELISA and Western blot
High protein-binding polystyrene 96-well plates (Corning) were coated overnight with 1 μg/mL recombinant proteins or 2 μg/mL chimeric peptide and in carbonate/bicarbonate coating buffer (pH 9.6). After washing in phosphate-buffered saline with 0.05% Tween-20 (PBST, Sigma-Aldrich), the wells were blocked with PBST containing 5% goat sera (Invitrogen) for a minimum of 1 h at room temperature (RT). Undiluted culture supernatant was incubated on coated plates at 50 μL/well for 1 h at RT. Mouse anti-His C-term antibody (Invitrogen) and expansion media were used as positive and negative controls. Plates were washed three times with PBST and probed for 1 h at RT with HRP-labeled goat anti-mouse IgG (Southern Biotech, Birmingham, AL), which was diluted 1:1000 in PBST and 5% goat sera. The original screen of chimeric peptide derived cultures was additionally probed with HRP-labeled goat anti-mouse IgM (Southern Biotech). Subsequent screens of chimeric peptide antibodies were probed with isotype-specific HRP conjugates. Plates were washed five times with PBST and developed for 10 min at RT with TMB substrate (Millipore, Temecula, CA). Development was stopped by the addition of an equal volume of 2.0 N sulfuric acid (VWR, West Chester, PA). Absorbance was measured at 450 nm with an automated spectrophotometer.
For Western blots, recombinant proteins were reduced and denatured with NuPage® LDS sample buffer and sample reducing agent (Invitrogen), and then heated for 5 min at 95°C. Twenty micrograms of protein were separated by electrophoresis on 2-D NuPage 4–12% Bis-Tris gels (Invitrogen) with NuPage MES SDS running buffer (Invitrogen) at 200 volts for 30 min. Proteins were transferred to nitrocellulose (Invitrogen) using NuPage transfer buffer (Invitrogen) plus 10% methanol at 30 volts for 90 min. Membranes were blocked overnight at 4°C with TBS-casein (Bio-Rad, Hercules, CA). Blots were assembled on a miniblotter 28 (Immunetics, Boston, MA). Non-diluted supernatant was loaded into individual slots and incubated for 1 h at RT, and then washed extensively with PBST. Mouse anti-His C-term antibody (Invitrogen) and expansion media were used as positive and negative controls. Blots were then incubated with alkaline phosphatase (AP) labeled goat anti-mouse IgG or IgM at 1:1000 in TBS-casein for 1 h at RT. Blots were extensively washed followed by development with Western Blue AP substrate (Promega, Madison, WI) for 20 min at RT.
Purification of recombinant 6x histidine-tagged proteins
Frozen pellets of HEK293 cells were thawed on ice and resuspended with 10 mL/g of 50 mM sodium phosphate (pH 8.0), 500 mM sodium chloride, 10 mM imidazole, and 10 mM β-mercaptoethanol in the presence of Complete EDTA-free protease inhibitor (Roche Diagnostics, Indianapolis, IN). Cells were disrupted by sonication on ice for three cycles of 30 s each. NP-40 alternative (Calbiochem, Gibbstown, NJ) was added to a final concentration of 1% v/v followed by clarification by centrifugation at 38,500 g for 40 min at 4°C. The clarified soluble lysate was decanted. For ETO B.003, the remaining insoluble pellet was resuspended with the above buffer plus 8 M urea without NP-40 alternative. After dounce homogenization, remaining insoluble material was removed by centrifugation and the urea-soluble lysate was decanted and processed similar to the soluble lysate. One milliliter of Ni-NTA superflow resin (Qiagen, Valencia, CA) was added to the lysate. The suspension was mixed by gentle end-over-end rotation for 2.5 h at 4°C. Resin was collected by centrifugation at 200 g for 5 min, resuspended with 5 mL of lysate, and then transferred to a 5 × 1 cm column. Upon packing of the resin, the column was washed at room temperature with 50 mM sodium phosphate (pH 8.0), 500 mM sodium chloride, and 10 mM β-mercaptoethanol in a step-wise imidazole gradient increasing from 10 to 30 mM in 3 × 10 mL wash steps. NP-40 alternative was included at 0.1% v/v in the first two wash steps. Histidine-tagged proteins were eluted with 6 × 1 mL fractions of 50 mM sodium phosphate (pH 8.0), 500 mM sodium chloride, and 300 mM imidazole. Insoluble B.003 ETO wash and elution steps included 8 M urea in the above buffers, but did not contain NP-40 alternative. Buffer exchange was performed at RT using a HiPrep desalting 26/10 column (GE Healthcare, Piscataway, NJ) into 50 mM sodium phosphate (pH 7.4) and 150 mM sodium chloride. Insoluble B.003 ETO buffer exchange included 6 M urea in the above buffer. Purity was assessed by SDS-PAGE followed by Coomassie staining with SimplyBlue™ Safe Stain (Invitrogen). Western blot analyses were performed by transferring proteins onto nitrocellulose using the iBlot system (Invitrogen). Histidine-tagged proteins were detected using an AP-conjugated C-terminal specific mouse anti-6xhistidine antibody (Invitrogen) at 1:2000 in blocking buffer followed by visualization using NBT/BCIP substrate (Promega).
Purification of monoclonal antibodies
Hybridoma culture supernatants were diluted 1:1 with binding buffer (50 mM boric acid, 4 M sodium chloride [pH 9.0) and passed through a 5 mL HiTrap MAbSelect SuRe™ protein A column (GE Healthcare) using the AKTAxpress chromatography system. The column was washed with 10 column volumes of binding buffer followed by elution with 5 column volumes of 50 mM sodium citrate (pH 3.0), 50 mM sodium phosphate, and 300 mM sodium chloride. Antibody eluates were stored in a sample loop and immediately buffer exchanged into 50 mM sodium phosphate (pH 7.4), 150 mM sodium chloride using a HiPrep 26/10 desalting column (GE Healthcare). Antibody containing fractions were pooled and 0.2 μm filtered through a PES Supor membrane syringe filter (Pall Life Sciences, Ann Arbor, MI). Immunoglobulin levels were quantified using A280 and purity was assessed using SDS-PAGE under reducing and non-reducing conditions.
Biotinylation of monoclonal antibodies
Antibody concentration was adjusted to 2 mg/mL with reaction buffer (100 mM sodium phosphate, 150 mM sodium chloride pH 7.2) to a final reaction volume of 500 μL. Immediately before use, 170 μL water was added to 2 mg NHS-PEO4-Biotin (Thermo Fisher Scientific, Rockford, IL). After resuspension, 6.7 μL of the resultant 20 mM biotin solution was transferred and mixed with the antibody solution to obtain a 20-fold molar excess of biotin to antibody molecules. The reaction was incubated at RT for 30 min. Non-reacted NHS-PEO4-Biotin was removed using Zeba™ spin desalting columns (Thermo Fisher Scientific) with 100 mM sodium phosphate (pH 7.2) and 150 mM sodium chloride according to the manufacturer's protocol. Biotin incorporation was verified using a HABA biotin quantitation kit (Thermo Fisher Scientific) according to the manufacturer's protocol.
Limit dilution cloning
Hybridomas were cloned by limit dilution into 2 × 96 well plates. Each well was microscopically observed to confirm the presence of a single cell. After incubation for 12 days, wells with growth originating from a single cell were screened by ELISA.
Expansion of hybridomas
Selected monoclonal hybridomas were expanded to 500 mL in cell culture bags (Lampire, Pipersville, PA) and allowed to grow to exhaustion (<20% viable cells). Supernatant was decanted and cells were pelleted by centrifugation. Clarified supernatants were passed through a 0.2 μm PES filter (Nalge Nunc, Rochester, NY) prior to purification.
Sandwich ELISA
Supernatant
Capture MAb supernatant was diluted 1:7.5 in coating buffer and 50 μL/well was coated onto 96-well ELISA plates (Costar, Corning, NY) overnight at 4°C. The plates were washed one time with PBST and wells were blocked with 300 μL 3% BSA/PBST for 1 h at RT. The blocking solution was aspirated and 50 μL of antigen, diluted to 1 μg/mL in 1% BSA/PBST, was added to each well for 1 h at RT. Detection MAb supernatant was diluted 1:7.5 in 1%BSA/PBST and incubated with horseradish peroxidase (HRP)-labeled goat anti-mouse IgG-Fc antibody (Bethyl, Montgomery, TX) at a final concentration of 150 ng/mL at RT for 30 min. Plates were washed three times with PBST. An equal volume of 3 mg/mL mouse IgG (dialyzed to remove sodium azide; BioCheck, Foster City, CA) was added to the MAb-HRP detector complex prior to transferring 50 μL to each well for 1 h at RT. Plates were washed three times with PBST followed by addition of 50 μL TMB substrate solution (Sigma) per well. The reaction was allowed to develop for 10–30 min and was stopped with 2 N sulfuric acid (VWR). Absorbance at 450 nm was read using a SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA). Signal to noise ratios were determined by dividing the OD of wells with antigen by the OD of wells with buffer for each antibody pair. Positive antibody pairs were identified by having signal-to-noise ratios > 2.0.
Purified biotinylated MAb
To test purified MAbs and purified biotinylated MAbs in sandwich ELISA, the following changes were made to the protocol described above for supernatant. Purified capture MAbs were coated directly onto ELISA plates at 2 μg/mL. Purified biotinylated detection MAbs were diluted to 1 μg/mL in 1%BSA/PBST and 50 μL were added to each well followed by incubation for 1 h at RT. After plates were washed three times with PBST, 50 μL of HRP-labeled streptavidin (Pierce, Rockford, IL), diluted 1:5000 in 1% BSA/PBST, were added to each well. The remainder of the assay was performed as previously described.
Results
Recombinant human ETO and AML1-ETO proteins
Recombinant proteins were expressed in HEK293 cells. The construct used for recombinant ETO antigens (Fig. 1) encoded a predicted 35 kDa molecular weight protein corresponding to AML1-ETO fusion protein amino acids 453–752 of GenBank accession D13979. Expression analysis revealed approximately 50% solubility (data not shown). Nickel affinity chromatography of the soluble HEK293 lysate yielded two major protein bands at 35 and 23 kDa, which were separated by size-exclusion chromatography and analyzed by SDS-PAGE (Fig. 2B) and anti-6x histidine Western blot (Fig. 2C). The identity of the 23 kDa protein, named ETO B.001, was assumed to be an N-terminal truncation product of the larger 35 kDa ETO B.002 protein due to reactivity of an anti-6x histidine antibody with the C-terminus of both proteins. To characterize ETO B.001 further, mass spectrometric analysis was performed on SDS-PAGE gel slices for each protein. Tryptic peptides were analyzed by nano LC/MS/MS followed by processing with the Scaffold™ algorithm (data not shown). Evidence for amino acids upstream of the sequence EEAVNE was not found in ETO B.001. The insoluble fraction of the HEK293 cell pellet was extracted with 8 M urea. Nickel affinity chromatography in the presence of 8 M urea yielded one major 35 kDa band, named ETO B.003, as shown by SDS-PAGE (Fig. 2B) and anti-6x histidine Western blot (Fig. 2C).
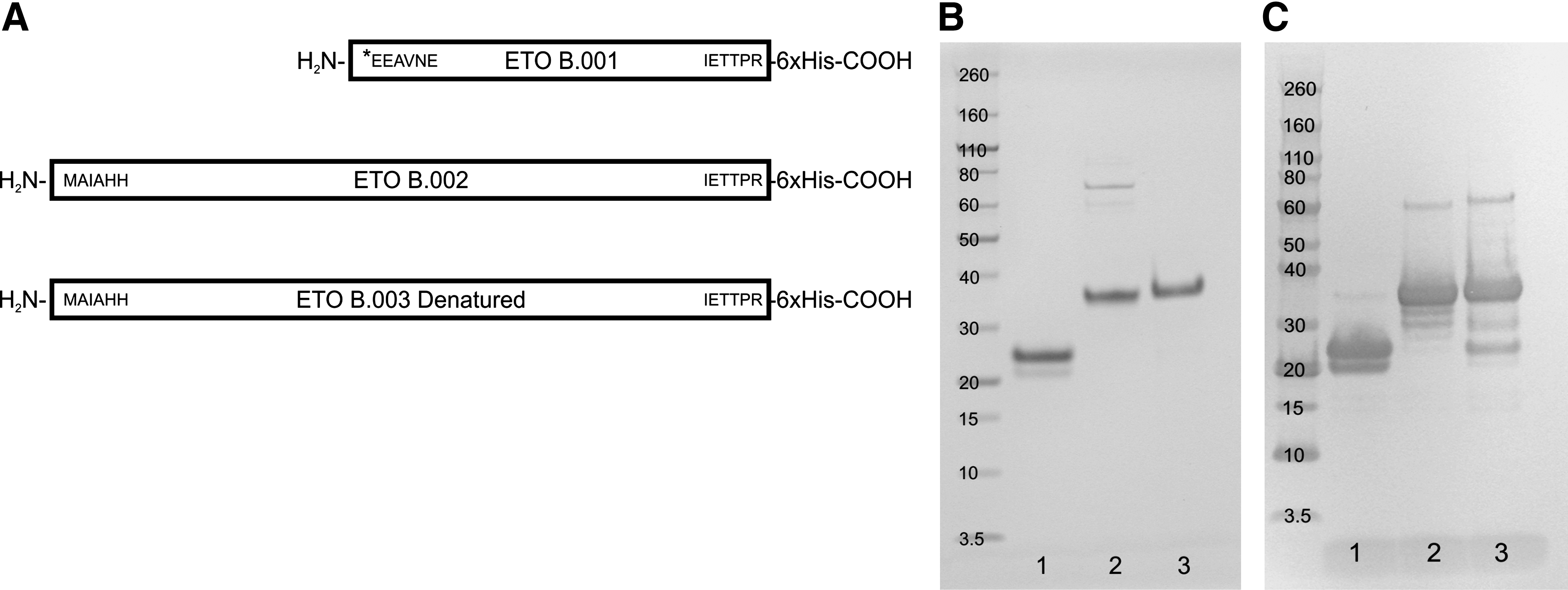
Purification of recombinant ETO proteins used for immunization. (
In contrast to the selected ETO peptide (Fig. 3A), an expression construct encoding the full-length 752 amino acid AML1-ETO fusion protein was used to transfect HEK293 cells. The expressed protein was extracted from the soluble portion of the HEK293 cell pellet and purified by nickel affinity chromatography followed by SDS-PAGE (Fig. 3B) and anti-6x histidine Western blot (Fig. 3C) analysis. Although the predicted 83 kDa protein was obtained, proteolysis was observed as shown by a protein “laddering” effect in SDS-PAGE and anti-6x histidine Western blot.
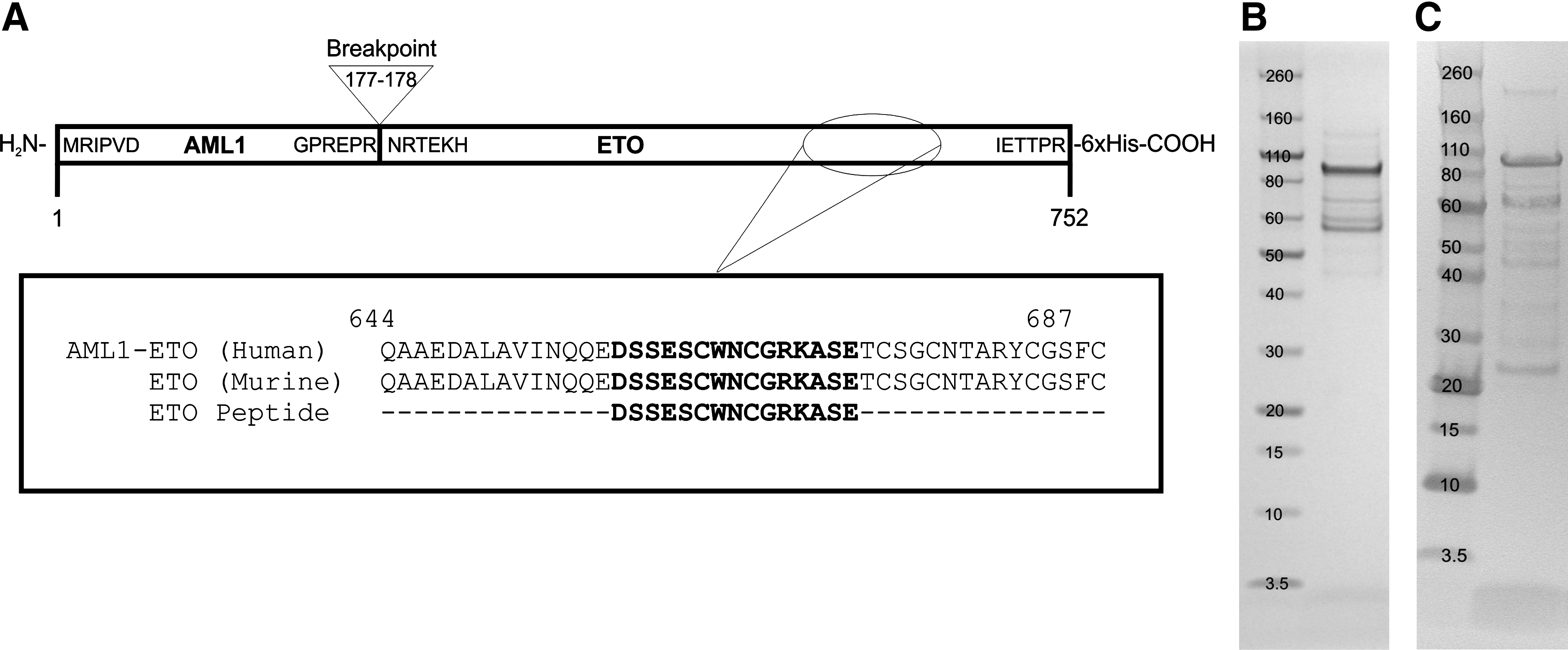
Breakpoint and amino acid analysis of the AML1-ETO fusion protein. (
Characterization of antibodies to recombinant human ETO
Balb/c mice were immunized with denatured ETO B.003 recombinant protein using RIMMS protocol. Lymphocytes were harvested from draining lymph nodes and fused with a mouse myeloma partner. Antibody containing supernatants from the resulting hybridoma cultures were tested in ELISA for reactivity to ETO B.001, ETO B.002, ETO B.003, an irrelevant 6x histidine-tagged protein, and the AML1-ETO fusion protein (Fig. 4A). Strong signals were obtained from 10/10 cultures when tested against ETO B.002 and ETO B.003, but only 3/10 cultures displayed slight reactivity to ETO B.001. Most of the cultures (8/10), displayed moderate reactivity with the AML1-ETO fusion protein. All of the tested cultures were negative for binding against an irrelevant 6x histidine-tagged recombinant protein. Cultures were also tested for reactivity against recombinant AML1-ETO fusion protein in Western blot (Fig. 4B). Positive reactivity was observed in 9/10 cultures at the same molecular weight as indicated by an anti-6x histidine antibody. The only negative culture, 6F8, interestingly exhibited the lowest ELISA signal. These results indicate that the epitope repertoire of this set of hybridomas is linear in nature and that recognition is clustered upstream of the sequence EEAVNE in our recombinant ETO proteins. Although ETO specificity was attained, this set of antibodies failed to pair with a previously generated set of AML1 antibodies in a sandwich ELISA assay using recombinant AML1-ETO as the analyte (data not shown).

Characterization of anti-ETO antibodies derived from recombinant ETO protein. (
Characterization of ETO peptide antibodies
In a second attempt to generate ETO antibody pairings with AML1 to allow detection of the AML1-ETO fusion protein, we identified an amino acid sequence near the C-terminus of ETO that was predicted to be soluble and solvent exposed by bioinformatics analysis. The selected ETO peptide was identified to be 100% homologous between mouse and human at the indicated position (Fig. 3A). To boost immunogenicity, a chimeric peptide was synthesized that contained a sperm whale myoglobin T-cell epitope and the ETO specific amino acids. Immunization of the peptide directly into the spleen, along with anti-CD40 agonist treatment, followed by isolation of spleenocytes and fusion to a mouse myeloma partner, resulted in a second set of ETO-specific antibodies. Hybridoma supernatants were tested in ELISA for reactivity to the immunizing ETO peptide, ETO B.001, ETO B.002, ETO B.003, an irrelevant 6x histidine-tagged protein, and the AML1-ETO fusion protein (Fig. 5A). Unlike antibodies raised against denatured recombinant ETO B.003, 5/5 of the ETO peptide antibodies displayed moderate to high reactivity with ETO B.001 in ELISA. Reactivity to the immunizing ETO peptide, ETO B.002, and ETO B.003 was strong in 5/5 cultures. All of the antibodies displayed moderate activity to the AML1-ETO fusion protein. To determine the isotype of the reactive antibodies, isotype specific HRP-tagged secondary antibodies were used in ELISA. The antibody isotype responsible for binding to the antigens was found to be IgG in 2/5 cultures, mixed IgG and IgM in 2/5 cultures, and IgM alone in 1/5 cultures.
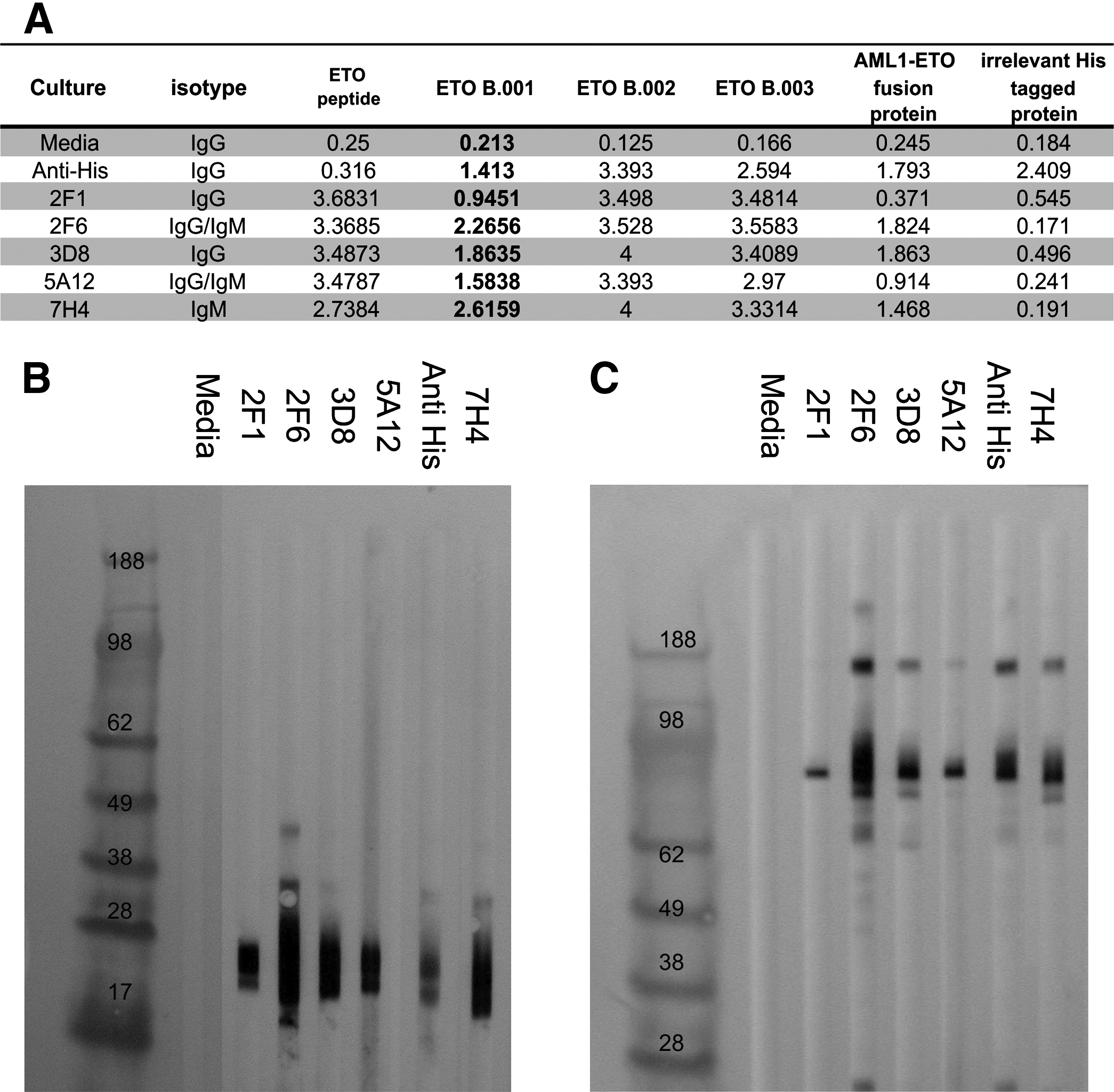
Characterization of anti-ETO antibodies derived from the chimeric ETO peptide. (
Hybridoma supernatants were also tested in Western blot against ETO B.001 and the AML1-ETO fusion protein (Fig. 5B). All of the ETO peptide raised antibodies were positive in Western blot with both ETO B.001 and the AML1-ETO fusion protein as indicated by bands at the same molecular weight as an anti-6x histidine antibody. These results show that the ETO peptide antibodies recognize a different set of epitopes when compared to antibodies raised against denatured recombinant ETO B.003 based on reactivity to ETO B.001 in ELISA and Western blot. The isolation of IgM hybridomas suggests that the method used for generating ETO peptide antibodies captured an early primary immune response.
Monoclonal antibody based sandwich ELISA for AML1-ETO
Polyclonal hybridoma cultures were cloned by limiting dilution. Resultant monoclonal IgG populations were expanded and immunoglobulin was purified by protein-A affinity chromatography. After identifying capture and detector antibody pairs, a sandwich ELISA was developed using a purified monoclonal AML1 specific capture antibody and a purified biotinylated monoclonal ETO peptide raised detector antibody. Recombinant AML1-ETO fusion protein, each ETO recombinant protein, and recombinant AML1 were tested by titration curve analysis (Fig. 6). At 125 ng/mL, the AML1-ETO fusion protein OD remained two times higher than wells containing no antigen. Signal to noise ratios increased with increasing fusion protein concentration. Cross-reactivity with fusion protein subunits such as the ETO proteins or AML1 was not observed, showing that the assay is specific for AML1-ETO fusion protein.
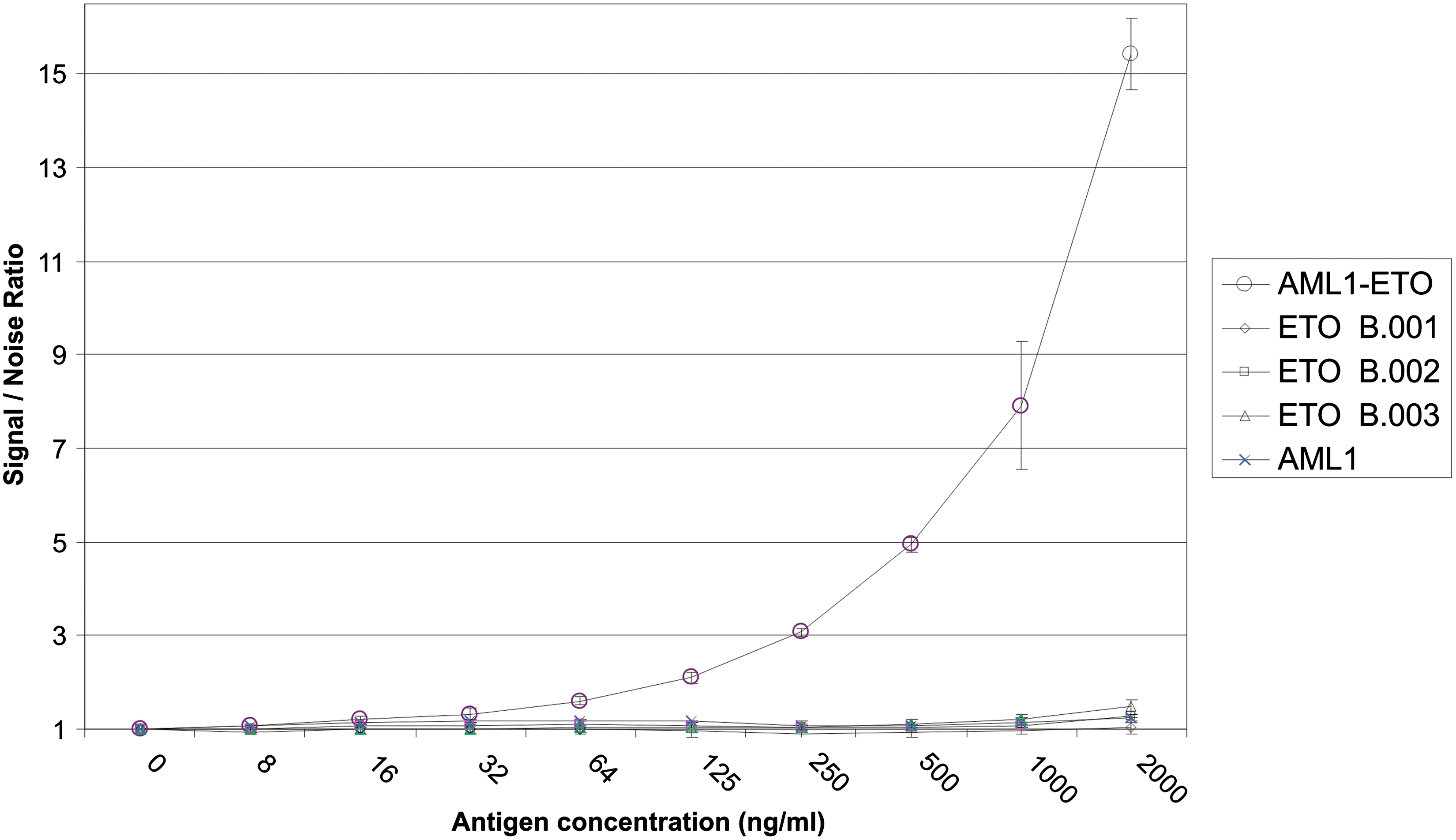
Antigen titration and cross-reactivity in AML1-ETO sandwich ELISA. Antigen was captured with MAb AML1 7G3.7. The antigens were titrated in 2-fold dilutions and detected with biotinylated MAb ETO 3D8.2. The assay was performed in triplicate. Signal-noise ratio was determined by dividing the OD 450 reading for antigen by the OD 450 reading for no antigen. This pair of antibodies is specific for the AML1-ETO fusion antigen and does not detect the individual antigens.
Discussion
The primary focus of this experiment was to develop ETO-specific MAbs. Our overall objective was to develop MAbs that would capture and detect the leukemia-associated transcription factor AML1-ETO. Our strategy was to develop specific antibodies to AML1 and ETO, respectively, located near the amino and carboxy termini of the AML1-ETO fusion protein. Use of these antibodies in a sandwich ELISA would facilitate detection of the AML1-ETO fusion protein, but not the individual protein subunits generated from the non-rearranged copies of each gene.
ETO B.001, B.002, and B.003 were purified from HEK-transfected cells under native or denaturing conditions. Although encoded for by the same recombinant vector, the size of ETO B.001 differed from that of ETO B.002 and ETO B.003. The predicted molecular weight of full length ETO is 35.2 kDa. Suspected proteolysis of the 35.2 kDa ETO B.002 protein resulted in a cleavage product of about 23 kDa, which was named ETO B.001. LC/MS/MS confirmed both bands were ETO. In addition to proteolysis, a major difference was seen in the solubility of the ETO proteins migrating at 35.2 and 23 kDa. A portion of ETO protein migrating at 35.2 kDa remained insoluble when extracted under native conditions, whereas the smaller ETO 23 kDa protein was completely solubilized in the same conditions.
Using the RIMMS protocol, and immunizing with ETO B.003, we were successful in developing a number of ETO and AML1-ETO specific antibodies. This was a mild surprise due to the highly conserved nature of ETO. ETO B.003 was derived from the 310 amino acid sequence at the C-terminal end of the ETO protein. This sequence has only three amino acid changes between human and mouse. In contrast to the RIMMS protocol, conventional immunizations with ETO B.003 were unproductive (data not shown).
ETO B.003 RIMMS derived cultures were negative for binding with ETO B.001 in ELISA and Western blot analysis. These anti-ETO antibodies did not complement an internally developed anti-AML1 clone 7G3.7 antibody in pairs testing with AML1-ETO fusion protein, although the anti-AML1 antibody demonstrated robust binding with the full length AML1-ETO fusion protein in ELISA and Western blot (data not shown). The inability to capture or detect AML1-ETO with this set of antibodies may reflect fusion protein folding such that epitopes derived from ETO B.003 are no longer surface exposed during sandwich ELISA testing.
We reasoned that epitopes in the more C-terminal ETO B.001 protein would have a greater chance of being surface exposed in the context of the AML1-ETO fusion protein. Antibodies to these epitopes would therefore have a greater chance of pairing with our existing AML1-specific reagents. Immunizations with recombinant ETO B.001 did not result in positive hybridomas. We identified a 15 amino acid potential B-cell epitope sequence near the C-terminal end of ETO. The hydrophilic nature of these amino acids suggested that the sequence may be surface exposed and a possible epitope for detector antibody binding in a sandwich ELISA assay. However, this region of human and mouse ETO is 100% homologous. Methods describing anti-self polyclonal responses using recombinant protein,(11) peptide-conjugated virus-like particles,(9) and MAP-conjugated peptides(10) prompted us to consider a chimeric peptide as an immunogen.
The ETO B.003 results described herein, and the highly conserved nature of ETO, all suggested that T-cell epitopes were the obstacle in generating ETO B.001-specific reagents Antibody responses against defined regions of proteins or conjugated peptides require simultaneous T-helper cell recognition with another part of the same protein or conjugated peptide. Polyclonal responses have been described after immunization with chimeric linear peptides containing both a foreign T-cell epitope fused to a B-cell epitope.(21,31) We chose the sperm whale myoglobin sequence FISEAIIHVLHSR(22) as a foreign T-helper cell epitope. This immunization peptide was synthesized as an N-terminal sperm whale T-helper cell epitope fused to a C-terminal ETO B-cell epitope.
The chimeric peptide was injected intrasplenically on three occasions over 11 days to traffic and focus rare immunoreactive B-cells into one anatomical location. On day 10 the mouse was pretreated with an anti-CD40 agonist antibody to expand peptide-specific B-cells to levels that could be immortalized during day 13 fusion. Five mixed isotype hybridomas were developed. All were reactive with the chimeric peptide immunogen, ETO B.001, B.002, B.003, and AML1-ETO in ELISA. In addition, all were reactive with ETO B.001 and AML1-ETO in Western blot analysis.
ETO-peptide derived hybridomas represent a mixture of IgM and IgG isotypes. We speculate that IgM represents a primary humoral response. In previous studies, treatment with CD40 agonist restored humoral responses to adenovirus in CD154-deficient mice.(29) CD40-CD154 signaling mediates up-regulation of inducible CD80/CD86 on APC.(32) The CD28-CD80/86 pathway, in the absence of CD40-CD154 co-stimulation, restores both Th2 activation and IgG humoral responses to adenovirus in CD154-deficient mice.(33) The IgG response to the chimeric ETO-peptide immunization may therefore consist of classical CD40-CD154 signaling with memory B-cell development and exogenous CD40 agonist-mediated up-regulation of CD80/86 on APCs. The end result is isotype class switching to IgG by prevention of T-helper cell anergy mediated by CD28-CD80/86 co-stimulation.(34)
The anti-ETO hybridoma 3D8, derived from peptide immunizations, was cloned and the purified monoclonal antibody was biotinylated. This MAb was able to detect AML1-ETO as low as 8 ng of fusion protein in a sandwich ELISA with an internally developed AML1-specific MAb used as a capture antibody. These experiments were performed in triplicate and not optimized for sensitivity. In other studies, simple conversion from a colorimetric to chemiluminescent ELISA has demonstrated an increase in sensitivity of 12–29 fold for cytokine detection.(35) Techniques to optimize reagent and buffer concentrations, as well as analyte pre-concentration, would be expected to further increase sensitivity. It is not unreasonable to predict a limit of detection below 1 ng after product development efforts.
Although MAbs specific to linear peptides of complete amino acid homology were generated, they may not be equivalent to anti-self protein antibodies. Linear amino acid peptide sequences used as immunogens may be modified compared to the native protein. Post-translational modifications and folding may influence linear epitope surface exposure. Kasumi-1 cell lysates expressing AML1-ETO(36) and mouse cell lines expressing ETO will be used in future studies to confirm anti-self protein specificity.
The efforts described here are the first step in detection of AML1-ETO protein from clinical samples. Identification of an AML1-ETO t(8,21) is important as these patients have a better prognosis after treatment with cytarabine.(37) Standard cytogenetic analysis (SC) is often the first confirmation of AML1-ETO t(8,21) at diagnosis. Although SC has high specificity, the sensitivity of SC is low because few actively dividing cells are analyzed and abnormalities present in rare cells are not detected.(38) Molecular diagnostic testing increases the limit of detection to 1% of neoplastic cells using FISH,(38) and RT-PCR is purported to detect down to 10−5 to 10−6 neoplastic cells.(39,40) This high degree of sensitivity with RT-PCR allows the detection of fusion protein transcripts from patients with normal-karyotype AML at diagnosis,(41) patients in long-term remission,(42) and even healthy adults.(43) The significance of these subclinical populations of malignant cells from different clinical pathologies remains to be determined. The ability to capture and detect AML1-ETO protein at diagnosis and during remission may provide clues to understanding the relationships among fusion protein transcript, protein, and leukemic disease. In addition, direct detection of the fusion protein may facilitate monitoring of disease progression and response to therapy.
In conclusion, we report the development of ETO and AML1-ETO-specific MAbs. These were derived after intrasplenic immunization with a chimeric peptide containing a foreign T-cell epitope fused to the B-cell epitope in conjunction with exogenous CD40 agonist treatment. One anti-ETO MAb detects recombinant AML1-ETO in a sandwich ELISA assay when used with a previously developed AML1-specific MAb. Efforts to determine clinical relevance of these reagents are in progress. These reagents may help understand the relationship between AML1-ETO fusion protein and AML t(8,21) leukemia. This immunization strategy may provide a unique mechanism to develop MAbs reactive with highly conserved proteins.
Footnotes
Author Disclosure Statement
The authors have no financial interests to disclose.