
Abstract
The secreted Mycobacterium tuberculosis (MTB) proteins, Ag85B and Hsp16.3, have been the focus of intensive research in recent years. These proteins have high sensitivity in bacterium-negative tuberculosis (TB) patients, and are valuable for the rapid diagnosis of bacterium-negative TB. Fusion proteins including multiple antigens such as Ag85B and Hsp16.3 provide improved sensitivity and specificity for serological diagnosis of active TB compared with a single antigen. Many studies have shown that the production of MAbs recognizing a specific repertoire of M. tuberculosis antigens and the tests based on monoclonal antibodies have been found to be valuable in positive detection of TB, particularly for smear-positive pulmonary TB. A number of MAbs are currently used for serodiagnosis of TB. Therefore, an Ag85B-Hsp16.3 fusion protein was expressed and purified using an E. coli system in this study. Three Ag85B-Hsp16.3 fusion protein-specific MAbs were generated by routine murine hybridoma techniques. The titer, specificity, and relative affinity of all three MAbs were determined by ELISA and the serological responses were analyzed. The levels of antigens in a proportion of TB patients were shown to be significantly higher than those in healthy controls. The sensitivity and specificity of the currently available detection systems is likely to be improved by the employment of a combination of these MAbs with others that are already in use.
Introduction
The 30 kDa major secretory protein of M. tuberculosis (Ag85B) is the most abundant protein exported by M. tuberculosis, as well as being a potent immunoprotective antigen and, consequently, a leading drug target.(2) Hsp16.3 is a small heat shock protein from M. tuberculosis, which was identified as a major membrane protein localized to the thickened cell envelope and is predominantly expressed under oxidative stress. Gene knock-out studies indicate that the Hsp16.3 protein is required for the replication of TB in host macrophage cells.(3) This protein is considered to be a signature of dormant M. tuberculosis.
The Ag85B-Hsp16.3 fusion protein used in this study has been purified and analyzed previously. Antibody responses to this fusion protein were detected with high sensitivity by ELISA in TB patients (data not shown). However, the use of multiple antigens, either individually or as fusion polyproteins, for the detection of humoral responses has limited specificity and sensitivity.(4,5) The detection of circulating Mycobacterium antigens using specific monoclonal antibodies (MAbs) has been shown to be a promising approach for the detection of active infection. Furthermore, the tests based on MAbs recognizing a specific repertoire of M. tuberculosis antigens was shown to increase the rate of detection of bacterium-negative TB. In this study, the Ag85B-Hsp16.3 fusion protein, expressed and purified from a prokaryotic system, was used as the antigen to generate a monoclonal antibody that may be valuable for the diagnosis of TB.
Materials and Methods
Materials
MTB strain H37Rv was kindly provided by Shaanxi Provincial Institute for Tuberculosis Control (Xi'an, China). Escherichia coli DH5α cells, the Sp2/0 cell line, and BALB/c mice (female, aged 6−8 weeks) were provided by the Laboratory Animal Center of the Fourth Military Medical University (Xi'an, China). Ni2+-NTA purification kits were purchased from Invitrogen (Carlsbad, CA); the SBA Clonotyping System was purchased from Southern Biotech (Birmingham, AL); anti-16-kDa antigen MAb and anti-Ag85B rabbit polyclonal antibody were purchased from Abcam Company (Shanghai, China); HRP goat anti-mouse IgG antibody, HRP goat anti-rabbit IgG, and HRP goat anti-human IgG were purchased from Dingguo Biotech (Beijing, China). TB patient and volunteer blood samples were obtained from Wuwei Municipal Center for Disease Control (Gansu Province, China).
Target gene amplification
Primers were designed according to the nucleotide sequences of Ag85B and Hsp16.3 genes of MTB H37Rv strain. The target gene fragments were amplified using the genomic DNA of MTB H37Rv strain as a template. The Ag85B gene was amplified using the primer pairs: 5′-CGGATCCACCGCGGGCGCGTTCTC-3′ (containing the initiation codon) and 5′-CGAAGCTTGCCGGCGCCTAACGAAC-3′ and the Hsp16.3 gene was amplified using the primer pairs: 5′-GA
Expression, purification, and identification of the Ag85B-Hsp16.3 fusion protein
The recombinant plasmid pProEX HTa-Ag85B-Hsp16.3 was transformed into E. coli DH5α cells and expression induced by isopropyl-1-thio-b-D-galactopyranoside (IPTG) (0.6 mM) at 37°C. Following induction, bacteria were pelleted at 12,000 rpm at 4°C for 2 min. The expressed protein was resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE, 10% gel) and the identity confirmed by Western blotting with monoclonal mouse anti-Ag85B and monoclonal mouse anti-Hsp16.3 antibodies. The fusion protein was purified by affinity chromatography using a Ni2+-NTA purification system under denaturing conditions. The urea concentration in the solution was gradually decreased allowing spontaneous renaturation of the protein. The purified protein was first dialyzed overnight at 4°C against buffer containing 6 M urea. The protein was dialyzed further against buffers containing 4, 3, 2, 1, and 0.5 M urea, with a final dialysis against PBS buffer. Each dialysis was carried out for at least 4 h. The expressed protein was separated by SDS-PAGE (12% gel) and the identity confirmed by Western blot analysis using polyclonal rabbit anti-Ag85B (1:1000) and monoclonal anti-16-kDa antigen (1:3000) as the primary detection antibodies.
Preparation of MAb cell line, screening, and purification of ascitic fluid containing MAbs
Female BALB/c mice (aged 6−8 weeks) were inoculated with purified Ag85B-Hsp16.3 fusion protein (50 mg/mouse) emulsified with an equal volume of complete Freund's adjuvant (Sigma, Ronkonkoma, NY) via subcutaneous injection and boosted on day 14 with incomplete Freund's adjuvant (Sigma) emulsion via a similar route. A second boost (50 mg/mouse) was administered without adjuvant intravenously via the tail vein on day 28. Ten days after the third immunization, mice were bled from caudal vein and the serum titer was determined by indirect ELISA. The mouse with the highest antibody response was sacrificed, and the spleen cells were fused with non-immunoglobulin producing Sp2/O murine myeloma cells at a ratio of 10:1 in serum-free medium using polyethylene glycol as the fusion agent. Monoclonal cell lines were established by limiting dilution of the colonies from positive wells. The resulting hybridoma cells were mixed with serum-free media to adjust cell density to 1−2×109 cells/L and 0.5 mL injected into mice intraperitoneally. Ascitic fluid was collected from the abdomen 10 to 15 days after the injection and centrifuged at 3000 rpm for 15 min. Supernatants were collected and MAbs purified by caprylic acid-ammonium sulfate precipitation.
Determination of MAb titer by indirect ELISA
A 96-well plate (NUNC, Nagel, Roskilde, Denmark) was coated with 10 μg/mL of protein and then blocked with 10% fetal bovine serum for 1 h to reduce non-specific adsorption. Serial dilutions of the purified antibody were added and incubated at 37°C for 1 h. HRP-conjugated goat anti-mouse antibody was used as the secondary antibody and color was developed with TMB substrate (Sigma). Supernatants from Sp2/0 cell culture and serum from the immunized mice were used as negative and positive controls respectively.
Western blot analysis of MAb specificity
Expressed proteins (Ag85B and Hsp16.3) were resolved by SDS-PAGE (10% gel) and then transferred onto nitrocellulose membrane. Fat-free milk (50 g/L) was used to block the membrane at 37°C for 1 h. The membrane was then incubated with 10 μg/mL of the purified MAb (E6E4H12) in a 37°C shaker for 1 h. The membrane was washed with PBS three times before incubation with the HRP-goat-anti-mouse IgG detection antibody (1:5000 dilution) in a 37°C shaker for 1 h. The membrane was then washed with PBS three times and visualized by enhanced chemiluminescence according to the manufacturer's protocol (Amersham Biosciences, Piscataway, NJ).
ELISA assay to evaluate MAb specificity and cross-reactivity
A 96-well plate was coated with target proteins at the concentration of 10 μg/mL (Ag85B, Hsp16.3, and Ag85B-Hsp16.3) and unrelated His-tag fusion proteins at the concentration of 10 μg/mL (Rpf, ESAT6-CFP10, RpfE, and Hsp65). The cloning, expression, and purification of Ag85B, Hsp16.3 Rpf, ESAT6-CFP10, RpfE, and Hsp65 were performed in this laboratory as described previously.(6,7) Plates were incubated with the purified MAb (10 μg/mL) as the primary antibody for 1 h at 37°C. The plates were washed three times and incubated with the HRP-conjugated sheep anti-mouse IgG (1:5000 dilution) secondary antibody. The plate was washed again, developed with TMB substrate, and the absorbance value at 450 nm was measured.
ELISA assay to identify MAb class and subclass
The immunoglobulin class and subclass of each MAb was determined using the SBA Clonotyping System according to the instructions provided by the manufacturer.
Determination of the relative MAb affinity
A 96-well immunoplate was coated with the Ag85B-Hsp16.3 fusion protein (10 μg/mL) at 4°C overnight, and was then blocked with 10% fetal bovine serum for 1 h. Serial dilutions of the purified MAb were incubated at 37°C for 1 h. The plate was rinsed and incubated with the HRP-conjugated goat anti-mouse antibody (1:5000 dilution) at 37°C for 1 h. After washing, TMB substrate was used for color development; the OD value was measured at 450 nm and in order to determine the relative affinity.
Analysis of serum samples
A 96-well plate was coated overnight at 4°C with 10 μg/mL of a selected MAb (E6E4H12). Serum dilutions (100 μL/well) were incubated for 1 h at 37°C. A total of 21 serum samples were selected, including 15 samples from smear or culture-positive patients and six samples from healthy volunteers. Serum was separated from blood samples immediately by centrifugation and stored at −70°C. The plates were washed three times and the polyclonal rabbit anti-Ag85B antibody (1:5000 dilution) was added. Plates were washed again and HRP goat anti-rabbit IgG was added (1:5000 dilution). After a further repetition of the washing steps, color was developed using TMB substrate.
Data management and statistical analysis
All data were entered into a Microsoft Office Excel file. The mean and standard deviation (SD) of the optical density values of individual groups were calculated. The cut-off value was defined as the mean OD value of the negative control sera plus three standard deviations (3 SD). Subsequently, individuals were scored as positive for the specific antibody response when the optical density value of the sample was equal to or exceeded the cut-off value. The difference between groups was analyzed using Student t tests.
Results
Expression, purification, and identification of the Ag85B-Hsp16.3 fusion protein
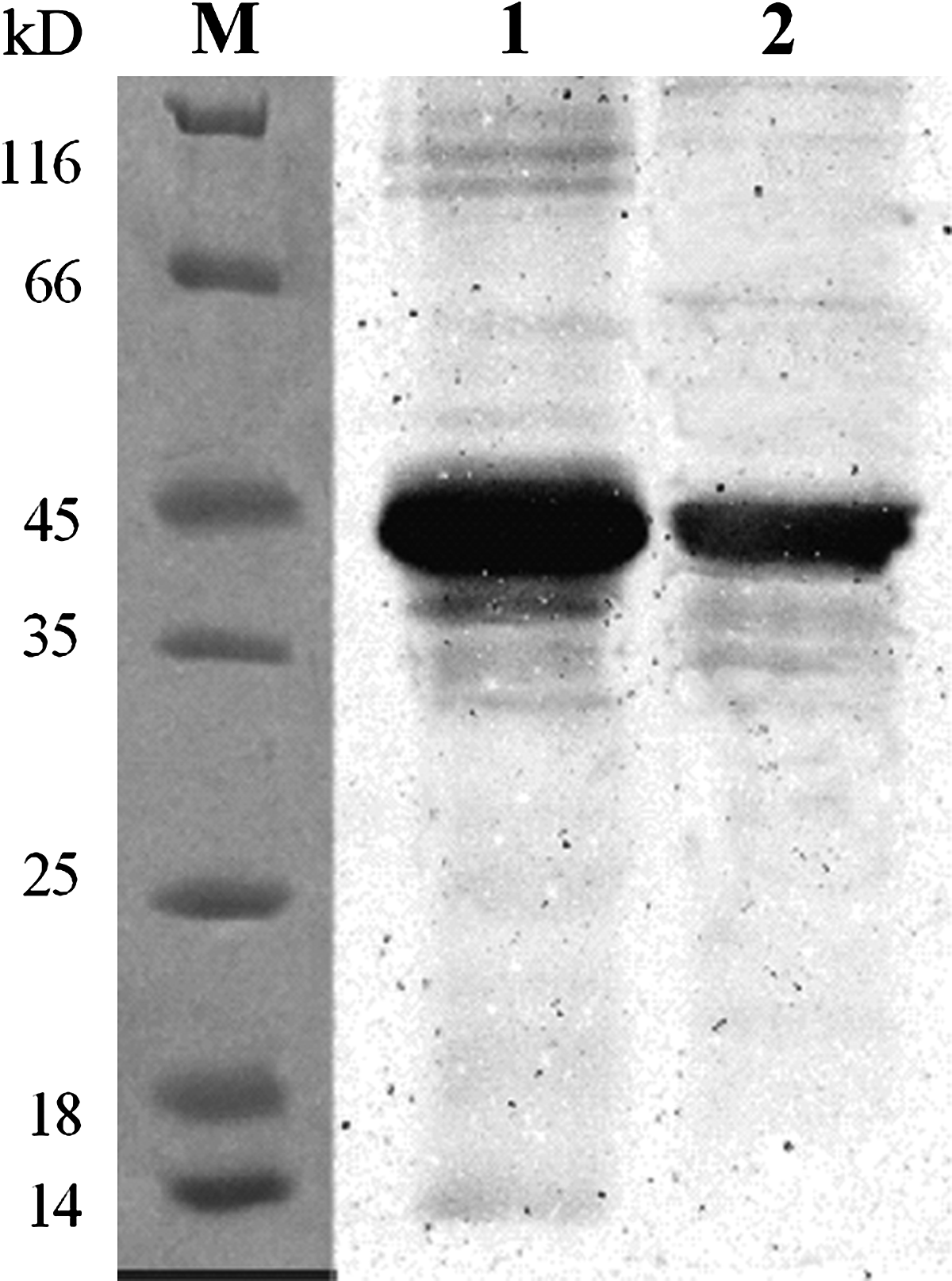
The expressed recombinant protein was identified with a relative fraction size of 49 kDa, approximately equivalent to the sum of the sizes of Ag85B and Hsp16.3. The Western blot assay indicated that the expressed products contained the binding site of Ag85B and Hsp16.3. The Ag85B-Hsp16.3 fusion protein was subjected to Ni2+-NTA purification under denaturing conditions and the purity of the protein was confirmed by SDS-PAGE (Fig. 1). A distinct protein band with a relative molecular mass (Mr) of 49 kDa was detected and the identity of the expressed fusion protein product was confirmed by Western blot analysis with Ag85B and Hsp16 specific antibodies (Fig. 2).

The expression of unpurified and purified Ag85B-Hsp16.3 fusion protein in E. coli was analyzed by SDS-PAGE (12% gel). Proteins were visualized by Coomassie blue R-250 staining. M, protein molecular weight marker; lane 1, pProEX HTa-Ag85B-Hsp16.3 product without IPTG induction in DH5α; lane 2, pProEX HTa-Ag85B-Hsp16.3 product induced in DH5α; lanes 3, 4, purified Ag85B-Hsp16.3.
Western blot analysis of expression product. Identification of proteins was confirmed by Western blot assay using anti-16-kDa antigen MAb and polyclonal rabbit anti-Ag85B antibody. M, protein marker; lane 1, expression product detected with polyclonal rabbit anti-Ag85B antibody; lane 2, expression product detected with anti-16-kDa antigen MAb.
Production and characterization of anti-Ag85B-Hsp16.3 protein MAb
The purified fusion protein was used to immunize BALB/c mice to generate an anti-Ag85B-Hsp16.3 monoclonal antibody. Three hybridoma cell lines that stably secreted MAb were obtained. The identity, class, subclass, and titer of each clone are listed in Table 1.
The isotypes of MAbs were identified by a SBA clonotyping system. The titers of MAbs in ascites were measured by indirect ELISA.
Specificity and cross-reactivity of the MAb
The monoclonal antibodies were shown to react specifically with the Ag85B-Hsp16.3 fusion protein and Ag85B by Western blot analysis (Fig. 3) and indirect ELISA (Table 2) while the unrelated His-tag fusion proteins (Rpf, ESAT6-CFP10, RpfE, and Hsp65) were not recognized. However, the Hsp16.3 protein was not recognized by the MAbs.
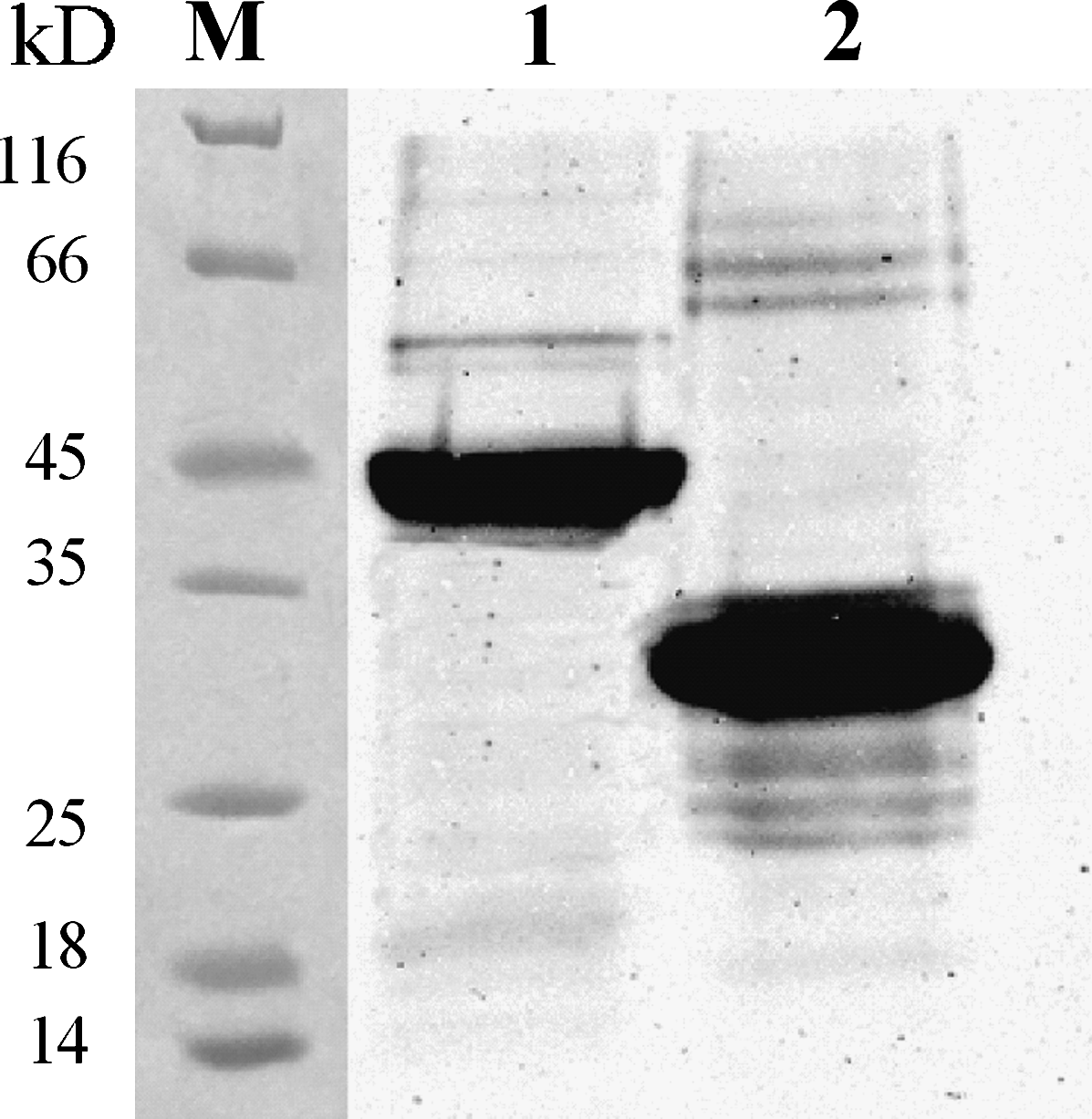
Western blot analyses of hybridoma supernatants. Antigen 85B and Ag85B-Hsp16.3 fusion protein were recognized by anti-Ag85B-Hsp16.3 fusion protein MAb (E6E4H12). M, protein markers; lane 1, Ag85B-Hsp16.3; lane 2, Ag85B.
Anti-Ag85B-Hsp16.3 fusion protein MAbs reacted with Ag85B-Hsp16.3 and Ag85B proteins but not Hsp16.3 protein or unrelated His-tag fusion proteins (Rpf, ESAT6-CFP10, RpfE, and Hsp65).
Investigation of MAb relative affinity
The relative affinities of MAbs were defined by the antibody concentration at which the OD value reached half the maximal signal at the plateau stage of antigen-antibody binding. The results showed that the order of relative affinity of the three selected MAbs was E6E4H12>A3B5C2>F6A12F6 (E6E4H12, 0.0001 mg/mL; A3B5C2,, 0.001 mg/mL; F6A12F6, 0.005 mg/mL (Fig. 4).
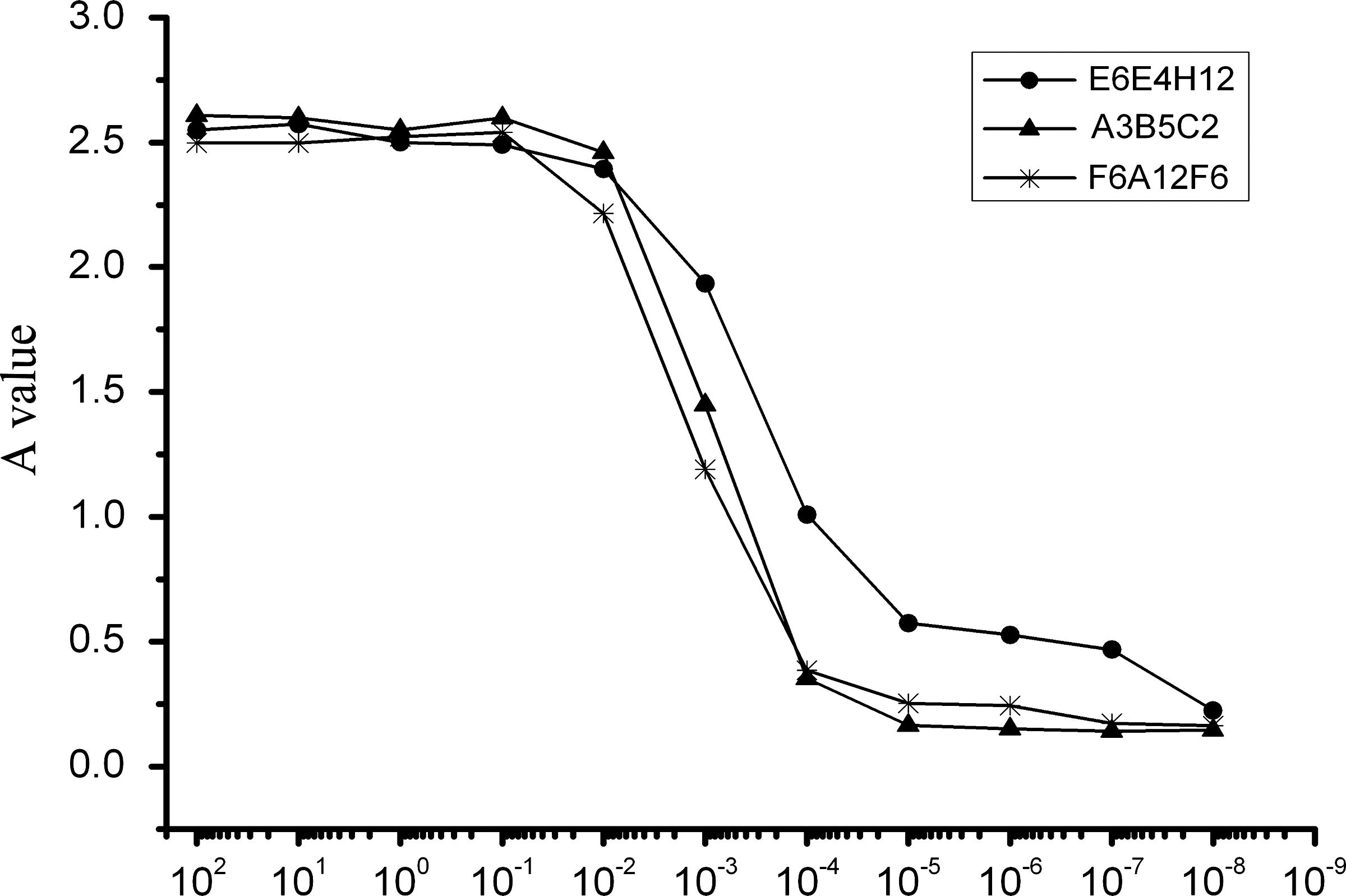
Relative affinities of anti-Ag85B-Hsp16.3 fusion protein MAbs for the Ag85B-Hsp16.3 fusion protein were analyzed by indirect ELISA assay of the serially diluted MAb. The relative affinity of MAb was represented as the antibody concentration at an OD value corresponding to 50% of the maximal signal obtained at the plateau stage of antigen-antibody binding.
Detection of serum
The levels of antigen detected by the MAb were measured by ELISA in 22 serum samples (Fig. 5). Antibody responses to the 22 serum samples from TB patients were analyzed using a cut-off value (mean value of healthy individuals plus 3 SD). The antigen was detected in serum samples from TB patients by the MAb with a sensitivity of 57.1% (8 of 14) and a specificity of 100%. The levels of antigen against the MAb in TB patients were significantly higher than those in healthy controls (p>0.002).
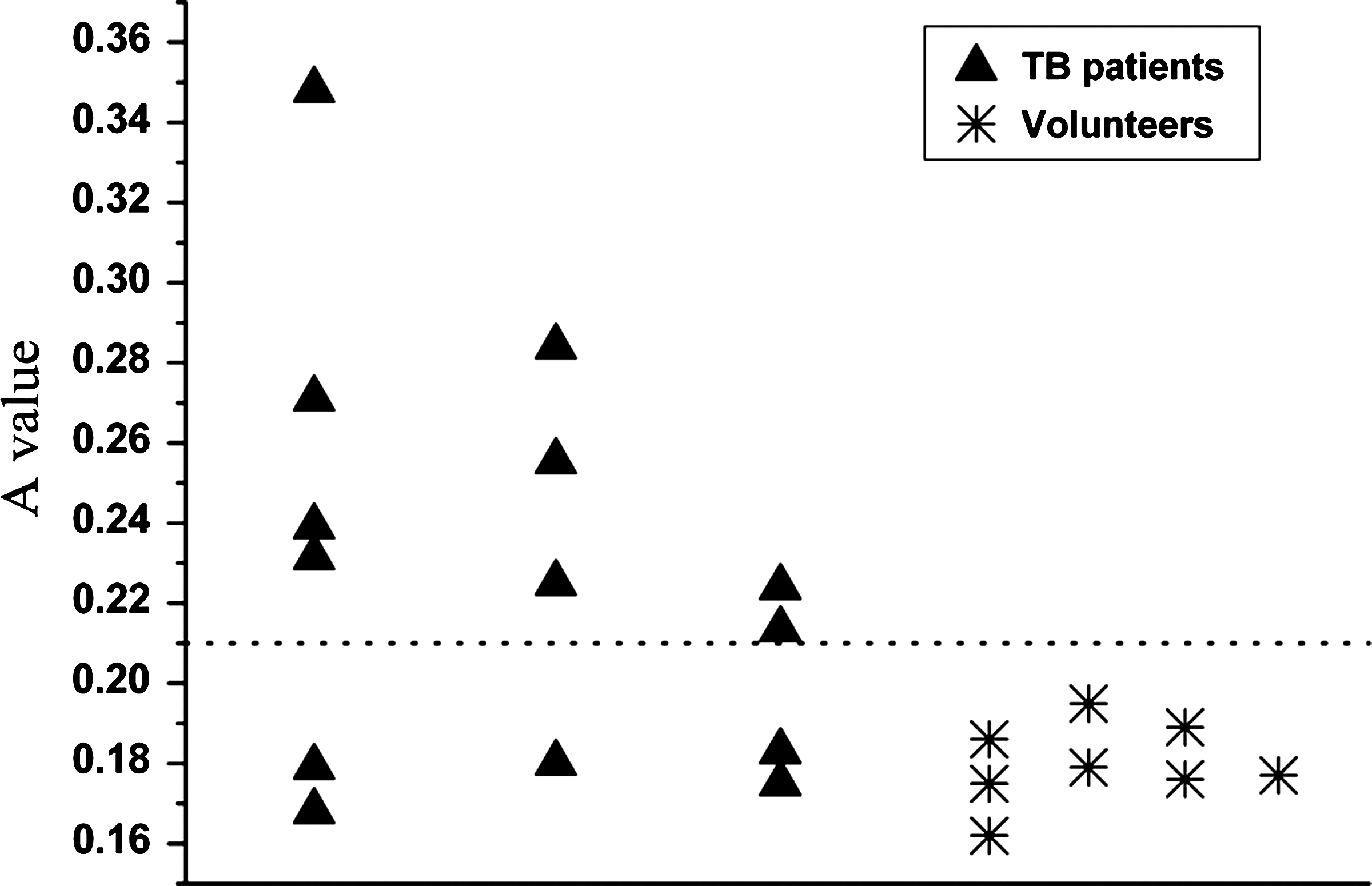
Levels of serum antigen were detected by the anti-Ag85B-Hsp16.3 protein in TB patients and were analyzed by ELISA. Data shown are the mean values of individual subjects from three independent experiments. Sera from patients and healthy subjects were tested simultaneously. The horizontal line indicates the cut-off value obtained from negative control groups. The cut-off value for individual antigens is 0.216.
Discussion
The detection of M. tuberculosis specific human antibodies is an important detection aid in the diagnosis of TB, especially for bacterium-negative TB. The serodiagnostic potential of many MTB recombinant proteins (Hsp16.3, Ag85B, RpfE, Hsp65, ESAT6) has been evaluated previously in patients with active TB.(3) High titers of antibodies against the Ag85B and Hsp16.3 MTB antigens were detected in most patients, indicating the potential value of these proteins in the serodiagnosis of TB in the clinic. The antigen 85B is a strongly recognized T-cell antigen in the first phase of TB infection and contains numerous well-characterized epitopes.(8–10) Furthermore, protective efficacy of this protein has been demonstrated in animal models.(7,11) It has been well documented that Hsp16.3, a homologue of the mammalian eye-lens protein α-crystallin (16-kDa protein, hspX, α-crystallin protein, Acr), plays a key role in protecting essential proteins from being irreversibly denatured during the TB-persistence phase, although the mechanism of this action remains unclear. Hsp16.3 contains T-cell and B-cell epitopes and is characterized as a molecular chaperone that functions by preventing the aggregation of proteins under stress conditions.(7,12,13) This protein is a member of the α heat shock protein (HSP) family represented by the low-molecular-weight HSPs and α-crystallin (A and B), which have been shown to be important in the maintenance of bacterial long-term dormancy.(14)
Ag85B and Hsp16.3 are M. tuberculosis antigens secreted in different phases of infection. It is speculated that using a combination of these two antigens is likely to increase the sensitivity and specificity of detection of anti-MTB antibody responses. Hence, in this study, Ag85B and Hsp16.3 were cloned, sequenced, and expressed as a fusion protein in E. coli. To ensure correct folding of the two proteins, a sequence encoding a l6 amino acid hydrophobic peptide rich in glycine and serine was inserted between Ag85B and Hsp16.3. The degree of flexibility of this linker ensures that the two proteins function separately. This purified protein was specifically recognized by the anti-16-kDa antigen MAb and polyclonal rabbit anti-Ag85B antibody. Furthermore, it was demonstrated that the Ag85B-Hsp16.3 fusion protein was recognized by human antibodies with improved sensitivity and specificity for the serological diagnosis of active TB (data not shown), thus indicating that recombinant protein antigens can be used for the serodiagnosis of TB in the clinic. However, the direct application of purified MTB antigens for the detection of humoral responses has limited specificity and sensitivity. These antigens may share antigenic epitopes with the BCG vaccine and therefore detection of antibody responses may be masked by BCG vaccination. However, there are no M. tuberculosis antigens in healthy people; therefore MAbs recognizing a specific repertoire of M. tuberculosis antigens may increase the positive detection of bacterium-negative TB and reduce the rate of false-positive results. The sensitivity of serum antibody competition tests (SACT) have been shown to be increased by the inclusion of more MAbs, particularly for the analysis of smear-positive pulmonary TB.(15) Several immunoassays have been established for detection of MTB antigens in serum, sputum, and cerebrospinal fluid of tuberculous patients using polyclonal or monoclonal antibodies raised against different M. tuberculosis antigens.(16) Moreover, TB infection may be detected earlier by detection of antigens rather than by detection of antibodies.
In this study, three anti-Ag85B-Hsp16.3 fusion protein MAbs of high specificity and relatively high affinity were generated by routine murine hybridoma techniques. The MAbs did not recognize the Hsp16.3 protein, suggesting that the epitope recognized by this MAb is located on the surface of Ag85B while the Hsp16.3 epitope is concealed or that some antigenic epitopes were lost in the generation of the recombinant fusion protein. Further analysis of the serological responses of the anti-Ag85B-Hsp16.3 protein MAb demonstrated that the levels of antigen in some TB patients were significantly higher than those in healthy controls where no antigen was detected (0/8). However, determination of antigen levels using the MAbs cannot completely distinguish between TB patients and healthy controls due to multiple factors including the different status of TB patients, bacillary load, the impact of BCG vaccine or drug treatment, and others.
In summary, this study demonstrates that MAbs against the M. tuberculosis Ag85B-Hsp16.3 fusion protein may be a useful tool for the sensitive and specific diagnosis of active TB patients. However, cross-reaction of this MAb with the M. bovis strain BCG may occur; therefore further investigation is required to define the limitations of this MAb for serodiagnosis of TB in BCG-vaccinated populations.
Footnotes
Author Disclosure Statement
The authors have no financial interests to disclose.
Acknowledgments
This work was supported by grants from the High Technology Research Project of China (863 Program-2007AA02Z473, 2008ZX10003-013-3) and the National Natural Science Foundation of China (no. 30972767).