
Abstract
Increasing evidence implicates IgG autoantibodies against oxidized forms of low density lipoprotein (oxLDL) in the pathophysiology of atherosclerotic arterial disease. However, insufficient knowledge of their structure and function is a key gap. Using an elderly LDL receptor-deficient atherosclerotic mouse, we isolated a novel IgG3k against oxLDL (designated MAb LO1). LO1 reacts with copper-oxidized LDL, but minimally with native LDL. Further analysis showed that MAb LO1 also reacts in vitro with malondialdehyde-conjugated LDL (MDA-LDL), a known key epitope in copper-oxidized LDL preparations. By screening a phage library expressing single chain variable region antibodies (scFv), we selected an anti-idiotype scFv (designated H3) that neutralizes MAb LO1 binding to MDA-LDL. Amino acid substitutions between H3 and an irrelevant control scFv C12 showed that residues in the H3 CDRH2, CDRH3, and CDRL2 are all critical for MAb LO1 binding, consistent with a conformational epitope on H3 involving both heavy and light chains. Comparison of amino acids in H3 CDRH2 and CDRL2 with apoB, the major LDL protein, showed homologous sequences, suggesting H3 has structural similarities to the MAb LO1 binding site on MDA-LDL. Immunocytochemical staining showed that MAb LO1 binds epitopes in mouse and human atherosclerotic lesions. The MAb LO1-H3 combination therefore provides a very promising model for analyzing the structure and function of an individual IgG autoantibody in relation to atherosclerosis.
Introduction
Although IgG antibodies that react with oxidized LDL (oxLDL) can readily be detected in serum, relatively little is known about their precise epitope specificities and functions.(10,11) IgG anti-oxLDL in mice tend to be IgG1, IgG3, and IgG2a/c isotypes, while in humans they are predominantly IgG1 and IgG3.(12) Theoretically, distinct IgG anti-oxLDL isotypes may have quite different consequences for atherogenesis, depending upon their relative capacities to activate complement and ligate proinflammatory Fcgreceptors (FcγR).(13–18) The proinflammatory potential of IgG antibodies in atherosclerosis was supported by the marked protection from lesion development in LDL receptor-deficient mice lacking FcγRIII (CD16).(19) On the other hand, passive immunization with human IgG1 or mouse IgG2b monoclonal antibodies (MAb) has been shown to be protective.(20,21)
Antibodies that react with oxLDL antibodies probably recognize a range of epitopes, expressed at different levels of oxidative modification in plasma and in atherosclerotic lesions. Much of what we know about these antibodies has been discovered through the generation of MAb in mice, either following immunization(22–27) or occurring spontaneously in hyperlipidemic mice.(28,29) So far the spontaneously occurring autoantibodies that have been studied have been mostly, if not entirely, of the IgM isotype.(28,29) With the aim to better understand the fine specificity and function of naturally occurring IgG anti-oxLDL autoantibodies, we generated hybridomas from the spleen of an elderly LDL receptor-deficient mouse that had not been immunized. This report describes the characterization of one of these new antibodies, together with an anti-idiotype single chain variable region antibody (scFv) with which it specifically reacts.
Methods
LDL modification
Human LDL (Calbiochem, Beeston, United Kingdom) was dialyzed against PBS overnight at 4°C to remove EDTA. Following dialysis, copper-oxidized LDL (Cu-oxLDL) was prepared by incubating LDL with freshly prepared copper sulphate (10 μM) at 37°C for 24 h. The oxidization reaction was stopped by adding an excess amount of chelating resin Chelex 100 (Sigma-Aldrich, Poole, UK). The extent of LDL modification was estimated by the relative electrophoretic mobility of the LDL particles using pre-cast 0.6% agarose, 1.0% barbital buffer gels (Beckman Coulter, Fullerton, CA).
Malondialdehyde-conjugated LDL (MDA-LDL) was prepared as described by Palinski and colleagues.(30) Briefly, MDA was synthesized by acid hydrolysis of malondialdehyde bisdimethylacetal. Dialyzed human LDL was incubated for 3 h at 37°C with 0.5 M MDA at a ratio of 100 μL MDA/mg LDL. The extent of modification was estimated with a carbonyl assay.(31) Briefly, 0.1 mg of modified LDL was added to 500 μL of 4 mg/mL bovine serum albumin (BSA). 700 μL of 0.1% (wt/vol) 2,4-dinitrophenylhydrazine (DNP) in 2 M HCl was then added and incubated for 1 h at RT. Following the addition of 500 μL 30% trichloroacetic acid, the mixture was centrifuged for 5 min at 18,000 g at 4°C, and then left on ice for 30 min. The precipitate was then washed three times with ethanol/ethyl acetate (1:1, vol/vol). The pellet was finally dissolved in 1 mL of 8 M guanidine hydrochloride, 13 mM EDTA, and 133 mM Tris (pH 7.4) and the OD read at 365nm. The results were expressed as moles dinitrophenol (DNP)/mg of protein (mol/mg) using an extinction coefficient of 21 mM-1/cm−1. The synthesis and analysis of MDA conjugated to human serum albumin (HSA) were prepared using similar protocols.
Trypsinization of LDL was performed by adding 10 μL washed and diluted bovine pancreatic trypsin-conjugated agarose beads (60 mL beads in 100 μL of PBS, Sigma-Aldrich) to 0.5 mL of 342 μg/μL LDL. 45 μL aliquots were removed at intervals between 1 min and overnight, added to 3 μL of 1 mg/mL bovine pancreas-derived trypsin inhibitor (Sigma Aldrich) and centrifuged at 9100 g for 5 min. Control LDL was processed in the same way but without the addition of trypsin. The extent of LDL modification was confirmed by electrophoresis as above.
Hypochlorite modification of LDL (Calbiochem) was achieved by incubating LDL (1 mg protein/mL in PBS) with reagent-grade sodium hypochlorite (1 mM, Sigma-Aldrich) to a final concentration of 1 mg LD/mL of 1 mM hypochlorite solution for 24 h.
Hybridomas and selection of monoclonal antibody
Hybridomas were generated by fusing Sp2/0 myeloma cells with splenocytes from a one-year-old female LDL receptor-deficient mouse that had been fed a high fat diet from the age of 6 weeks old to give a serum cholesterol 25–30 mmol/L. Hybridoma culture supernatants were screened by ELISA for the presence of antibodies that bound with native LDL or oxLDL (each coated onto plates at 10 μg/mL). Hybridomas with differential reactivity to native LDL and oxLDL were then subcloned twice prior to further characterization. The isotypes of MAb were determined using a mouse MAb isotyping kit (IsoStrip, Roche Applied Science, Burgess Hill, UK). The hybridoma secreting an IgG3k isotype control MAb HK-PEG-1 (anti-influenza virus) was purchased from the European Collection of Cell Cultures (Porton Down, Salisbury, UK). MAb were purified from culture supernatant using a protein-G affinity chromatography column.
Enzyme-linked immunosorbent assay
Maxisorb 96-well plates (Nunc, ThermoFisher Scientific, Waltham, MA) were coated with 50 μL antigen/well at 4°C overnight. Non-adherent material was washed out, and then the plates were blocked with 2% BSA/PBS for 1 h at room temperature (RT). Appropriately diluted culture supernatant or purified MAb was added and incubated for 1 h at RT. Plates were then washed, and wells incubated with goat anti-mouse Ig (SouthernBiotech, Birmingham, AL) at 1:5000 dilution. After further washing, antibody binding was detected with 3,3’,5,5’-tetramethylbenzidine (Sigma), and the reaction was stopped with 0.5 M H2SO4. The optical density was then measured with a Synergy HT microplate reader (BioTek, Winooski, VT) at wavelength 450nm.
Variation in salt concentration was achieved by diluting MAb LO1 in different concentrations of NaCl ranging from 2.15 to 18 mM. To test the effect of altering pH, MAb LO1 was dialyzed into 0.1 M sodium acetate buffer using Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific, Rockford, IL) and then mixed with 0.1 M sodium acetate solutions prepared using combinations of 0.1 M sodium carbonate and 0.05 M sodium hydroxide solutions to produce working buffers of pH ranging from 3 to 11.
Equal loading of ELISA plates with native or modified LDL was confirmed in parallel wells using goat polyclonal anti-apoB (Abcam, Cambridge, UK). In the case of assessing MAb LO1 binding to immobilized scFv constructs, similar coating of wells with different constructs was checked by measuring binding of anti-myc MAb 9E10 (Sigma-Aldrich) in parallel wells.
Sequencing of MAb LO1 VH and VL variable regions
The MAb LO1 VH variable region was amplified from hybridoma mRNA by RT-PCR using a series of degenerate oligonucleotide primers, which had been prepared by Eurofin MWG Operon (Ebersberg, Germany). The sense primer was based upon the leader sequence,(32) and used in combination with a universal antisense constant region oligonucleotide(33,34) to amplify the leader, VDJ, and start of the constant region. The VL variable region sequence was obtained in a similar manner. PCR products were cloned into pCRII (Invitrogen, Paisley, UK) and transformed into TOP10 E. coli. Individual colonies were selected and DNA isolated by miniprep (Qiagen, Hamburg, Germany) prior to sequence analysis using an ABI 3730 automated DNA analyzer. The sequences were analyzed by comparison with the International MunoGeneTics information system (IMGT;
Isolation of anti-idiotype scFv antibody
The Tomlinson I library (Medical Research Council, Cambridge, UK) is a non-immunized human scFv phagemid library that was constructed in pIT2 (HIS6 myc tag) from VH (V3-23/DP-47 and JH4b) and Vk (O12/O2/DPK9 and Jk1). The diversified residues were based mainly on positions that are diverse in the primary repertoire and which are known from crystallographic studies to make contact with antigen. These are VH amino acids H50, H52, H52a, H53, H55, H56, H58, H95, H96, and H97; and VL amino acids L50, L53, L91, L92, L93, L94, L95, and L96. The total diversity of the library is about 1.4 × 108.(36,37) The functional scFvs bind both proteins A and L, and either one of these or the myc or 6 histidine tags can be used for detection, purification, or immobilization.
The phagemid-containing TG1 (T-phage resistant E. coli) cells were amplified in a 2xYT microbial medium (Sigma) containing ampicillin (100 μg/mL) and glucose (1% w/v) and grown with shaking at 37°C until the OD600 was about 0.4. The exponentially grown library-containing TG1 was then infected with KM13 helper phage and selected by ampicillin (100 μg/mL) and kanamycin (50 μg/mL; Sigma). The expanded phagemid library was then precipitated with PEG/NaCl (20% w/v polyethylene glycol 6000, 2.5 M NaCl) and titered on TYE plates (15 g Bacto-Agar, 8 g NaCl, 10 g Tryptone, 5 g yeast extract in 1 L dH2O) containing ampicillin (100 μg/mL) and glucose (1%).
A magnetic bead–based method was employed to select LO1 binding phage-scFv. This was carried out in three steps. Initially an aliquot of the library phage (5 × 1012 PFU) was pre-cleared of irrelevant binding phage-scFv by rotating with blank human anti-mouse magnetic beads (Invitrogen) for 1 h at room temperature. The unbound fraction was then further pre-cleared against human anti-mouse Ig beads preincubated with control IgG3k (1.5 μg/200 μL beads). For positive selection of LO1 binding phage-scFv, the unbound fraction was then transferred to magnetic beads coated with LO1 (1.5 μg/200 μL beads) and incubated rotating for 1 h at room temperature. Selected phages were then eluted with trypsin (1 mg/mL in PBS) for 15 min at RT, and used to infect logarithmic grown TG1 (OD600 about 0.4) by incubating at 37°C in a water bath without shaking. One tenth of the infected TG1 were then titrated in serial 10-fold dilution for phage titrating. The remaining infected TG1 were spread on a 15 cm × 15 cm TYE plates containing ampicillin (100 μg/mL) and grown at 37°C overnight. Phagemid-containing bacteria were scraped from the agar surface into 2xYT media containing ampicillin (100 mg/mL) and glucose (1%) and grown at 37°C with shaking until OD600 reached 0.4. The propagated phagemid was then rescued by MK13 helper phage and precipitated with PEG/NaCl (20% (w/v) polyethylene glycol 6000, 2.5 M NaCl). The phage produced was then titrated for the next round of selection. In total, three rounds of selection were carried out with a decreased concentration of LO-1 (1 μg/200 μL beads) used in rounds 2 and 3. Following incubation of human anti-mouse beads with LO-1, potential non-specific binding sites on the beads were blocked with 3%BSA/PBS. Beads were washed between stages with 0.05% Tween-20/PBS.
After each round of selection, 95 colonies on titrating plates were randomly chosen and grown in 100 μL 2xYT containing 100 μg/mL ampicillin and 1% glucose in microtiter plates. The ELISA plates were coated with LO1 or control IgG3 KH-PEG-1 (10 mg/μL in PBS) overnight at 4°C and were then blocked with 2% BSA/PBS for 2 h at RT. 10 μL PEG precipitated phage in 100 μL of 2% BSA/PBS was added into each well for 1 h of incubation. After three washes with PBS-Tween-20, the bound phage was probed with a horseradish peroxidase-conjugated monoclonal antibody that reacts specifically with the M13 phage major coat protein product of gene VIII, followed by TMB substrate. Phages giving an OD450 in MAb LO1 coated wells three times higher than those in the control IgG3 coated wells were considered to be specifically reactive with MAb LO1.
All clones positive in the ELISA screen were sequenced. Phagemid double-stranded DNA was purified by miniprep kits (Qiagen, cat# 27104) and subjected to sequencing on an automated ABI3730 sequencing system operated by Imperial Central Sequencing Service. The primers used for sequencing were: VH: CGA CCC GCC ACC GCC GCT G and Vκ: CTA TGC GGC CCC ATT CA (Eurofins MWG Operon, Ebersberg Germany). The data were analyzed with open source software eBioX (
As the TAG stop codon is suppressed in TG1 E. coli, both scFv and pIII are expressed. To produce scFv alone, selected phages were transferred to HB2151 (a non-suppressor strain), which was induced with 1 mM isopropyl β-D-thiogalactoside (Sigma). Antibodies were then isolated on Ni-NTA agarose (Qiagen) and eluted with imidazole (250 mM). Purity was confirmed by SDS-PAGE stained with Coomassie blue and a His-tag In-gel Staining kit (Invitrogen).
Generation of domain swaps between scFv
To construct the H3 VH/C12 VL and C12 VH/ H3 VL, the light chains of each construct were swapped. To release the light chain from the vector, purified DNA from both H3 and C12 scFv was digested with Xho1 (Promega, Madison, WI) and Not1 (Promega) in the following reaction: 5 μg DNA, 2 μL 10x buffer, 2 μL BSA (1 mg/mL), 2.5 U Xho1, 2.5 U Not1 adjusted to 20 μL with dH2O. Samples were incubated at 37°C for 1 h. Vector samples were subsequently treated with CIAP in the following reaction: 4 μL 10x buffer, restriction digested 20 μL DNA, 0.01 U CIAP, volume adjusted to 40 μL with dH2O. Samples were incubated for 1 h at 37°C. Fragments were separated on 1% agarose gels, and the appropriate bands gel purified before ligation using the following reaction: 4 μL 5x ligation reaction buffer, 45 fmol ends of insert, 15 fmol ends of insert DNA, and 0.1 U T4 ligase (Invitrogen), with the final volume adjusted to 20 μL with dH2O. After overnight incubation at 16°C, DNA was purified by phenol/chloroform/IAA extraction and ethanol precipitation. Purified, ligated DNA was transformed into TOP10 E. coli and selected on ampicillin (100 mμ/mL) plates.
Generation of mutant H3
Mutant constructs were created using the Site-directed Mutagenesis™ kit (Agilent Technologies, Santa Clara, CA) on the C12VL/H3VH domain swap background. Mutagenic primers were designed by hand as specified in the kit's manual for single and multiple mutations. Forward and reverse mutagenic primers contained the desired mutation and annealed to the same sequence on opposite strands of the plasmid carrying the C12VL/H3VH domain swap insert. The primers were designed to be 25–45 bases in length with melting temperature (Tm) below 78°C to prevent secondary structure formation. The Tm for each primer was estimated using the formula: Tm = 81.5 + 0.41 (%GC) − 675/N - %mismatch, where N = the primer length, and %GC and %mismatch are whole numbers. Optimally primers contained a minimum 40% GC content and ended in two G or C bases. The desired mutation was located in the middle with 10–15 bases of correct sequence on both sides. All primers were manufactured by Eurofins MWG Operon (Ebersberg, Germany). To ensure that the mutagenic primers functioned adequately through annealing and amplification at the correct temperature, 25 cycles of PCR were performed for each primer pair on vector dsDNA containing the C12VL/H3VH domain swap scFv sequence. The results were analyzed through gel electrophoresis (0.7% agarose) for the presence of a band approximately 5600 bp in size that represented the amplified construct with the desired mutation. Finally, the fidelity of the mutation was validated by sequencing. Mutant scFv were amplified and isolated as above.
Peptide synthesis
Peptides representing sequences of H3 were synthesized by Cambridge Peptides (Cambridge, UK). All peptides were purified by HPLC and quality controlled by mass spectroscopy.
Immunocytochemistry
Sections of aortic roots from wild-type (WT) and Ldlr-/- mice fed a low-fat diet to the age of 22 weeks were prepared as described previously.(9) Human carotid endarterectomy specimens were collected with consent and approval from Institutional and National Ethics committees. Specimens were transported and dissected on ice, with a transport time of under 30 min. The tissue was inspected and areas of classical morphology with fibrous cap, lipid core, and shoulder were identified and snap-frozen in liquid nitrogen and stored at −80°C. Then the sections were cryosectioned at −20°C, embedded in OCT, and fixed in isopropanol. Cryosections were equilibrated in double-distilled water, then PBS, blocked in 10% normal goat serum for 30 min, and incubated in 10 μg/mL biotinylated LO1 or biotinylated isotype control at 4°C overnight in a humidified chamber. Antibody was then decanted, followed by a rinsing in PBS. Bound antibody was detected with goat anti-mouse AlexaFluor 568 (Molecular Probes, Invitrogen, Paisley, UK) at 1:200 for 120 min at 21°C. TOPRO-3 (Molecular Probes, Invitrogen) was used as a nuclear counterstain for general inspection, and macrophages were detected by 1:100 anti-mouse CD68-AlexaFluor 488 (MCA1957, Serotec, Oxford, UK) or 1:50 anti-human CD68-FITC (green) (clone KP-1, F7135, Dako, Ely, UK). Sections were mounted in 80% glycerol/PBS and imaged by confocal microscopy (Zeiss LSM510 Meta, Jana, Germany) under standard settings as before.(38)
Results
Generation and characterization of MAb LO1
Hybridomas were generated using splenocytes from a one-year-old female LDL receptor-deficient mouse that had not been immunized. Of 950 clones screened, we identified only 10 IgG antibodies with differential reactivity with native versus Cu-oxLDL. From these, we selected an IgG3k MAb, designated LO1, for further characterization. As shown in Figure 1A and B, MAb LO1 reacted with Cu-oxLDL but only minimally with native LDL, and reactivity with Cu-OxLDL was inhibited by including 50 μg/mL Cu-oxLDL in the buffer. We then tested MAb LO1 reactivity with malondialdehyde (MDA)-conjugated LDL, which is known to be a prominent epitope in copper-oxidized LDL preparations.(30) As shown in Figure 1C, MAb LO1 showed increasing binding over time of LDL exposure to MDA, in parallel with carbonyl adduction (Fig. 1D). The failure of MAb LO1 to react with MDA conjugated to human serum albumin (not shown) signifies that it does not simply react with MDA as a hapten.
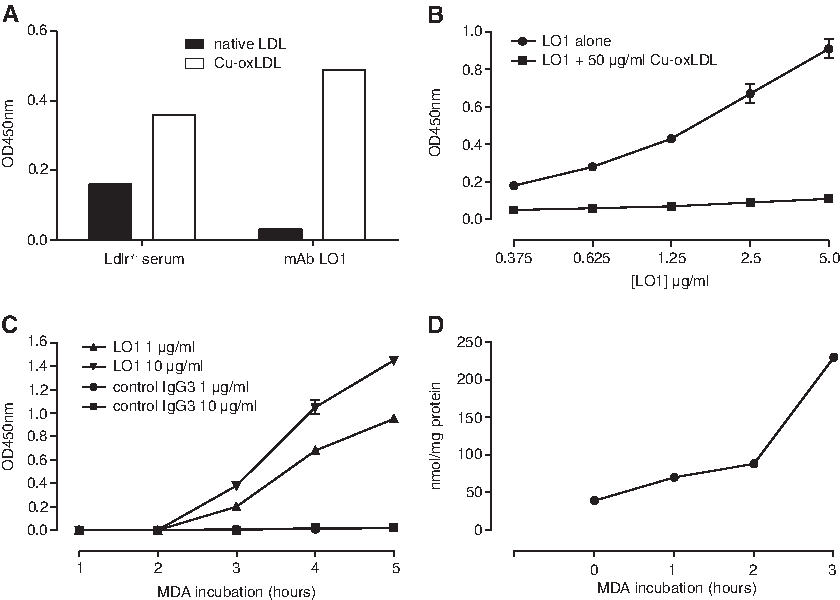
Initial characterization of MAb LO1. (
We checked whether LDL stored over 2 months in the absence of EDTA would become "automodified" in the presence of atmospheric oxygen to reveal the LO1 epitope and found that this was not the case (data not shown). MAb LO1 also failed to react with LDL incubated with hypochlorite for 24 h (not shown). Furthermore, trypsin treatment for up to 2 h did not reveal the LO1 epitope, despite clear evidence for proteolysis upon electrophoresis (not shown). Trypsin treatment of MDA-LDL for up to 2 h failed to influence MAb LO1 binding (not shown).
The immunoglobulin variable regions of the heavy and light chains of LO1 were sequenced and then examined by searching the IMGT database. The VH belongs to the VH1 family (V1-54) and is germline (Fig. 2). The VL belongs to the V6 family (V6–20) and is germline apart from minor differences.
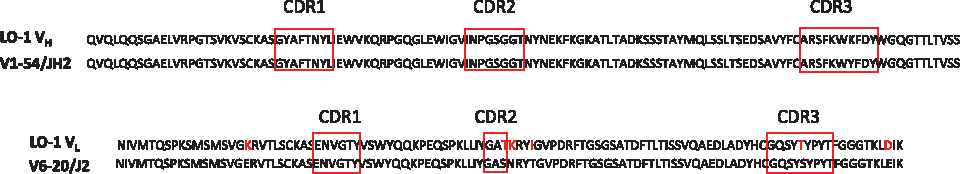
Amino acid sequence of the MAb LO1 VH and VL. The panel shows the MAb LO1 VH and VL sequences and their similarity to the germline V1–54/JH2 and V6–20/J2 sequences. LO1 amino acids that differ from germline are shown in red.
Generation of scFv anti-idiotype H3
Failure to Western blot MDA-LDL with MAb LO1 suggested that it reacts with a conformational epitope. In view of the challenge of defining a conformational epitope on a molecule as complex as LDL, we decided to explore LO1 binding further by developing an anti-idiotype antibody against the antigen binding site.(36,39,40) Affinity-purified MAb LO1 was used to select human scFv antibodies from an unbiased phage library. Three rounds of selection resulted in the isolation of a single scFv antibody, designated H3, which reacted with MAb LO1 but not with IgG3k control MAb HK-PEG-1 (Fig. 3A). Also shown in Figure 3A is the failure of MAb LO1 to bind a control scFv, designated C12, isolated from the same scFv library. As shown in Figure 3B, H3 but not C12 was able to neutralize the binding of MAb LO1 to MDA-LDL with an IC50 of 0.8–4.0 mg/mL, indicating that H3 binds MAb LO1 at or close to its recognition site for MDA-LDL. Changing the salt concentration had a very similar effect on binding of MAb LO1 to H3 as to MDA-LDL (Fig. 3C). There was also a similar profile of pH sensitivities with optimal binding at pH 6.0. Interestingly MAb LO1 binding to H3 had a slightly broader pH range compared to MAb LO1 binding to MDA-LDL (Fig. 3D).

Reactivity of anti-idiotype scFv H3. (
The CDR sequences of H3 were compared with the linear sequences of human and mouse apoB-100 using the Clustal W algorithm. As shown in detail in Figure 4, there are significant homologies between H3 CDRH2 and CDRL2 and peptide sequences of mouse and human apoB. Surprisingly, the relevant amino acids in H3 are situated not just among those that are diverse within the scFv library but also among constant determinants, particularly in the CDRL2 region.
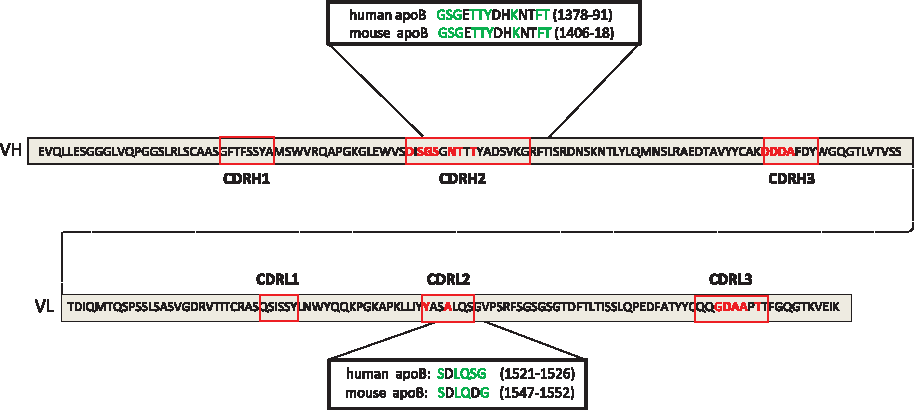
Similarities of sequences of LO-1 binding scFv and apoB-100; the figure shows the sequence of the VH and VL regions of scFv H3. Amino acids at each of the 18 points at which the Tomlinson I scFv library is diverse are in red. ApoB peptide sequences with similarities to H3 sequences are shown, with amino acids identical to H3 (green). Amino acid numbering is taken from NCBI protein sequences AAA51752.1 and NP_033823.2 for human and mouse apoB respectively.
As a prelude to site-directed mutagenesis of H3, we first swopped the VH and VL domains between H3 and C12. This established that the binding of MAb LO1 to H3VH chain paired with C12VL was similar to native H3, although the signal was slightly reduced at all concentrations of MAb LO1 (Fig. 5A). In contrast, no binding of LO1 occurred to a construct containing the H3VL chain paired with the C12VH (Fig. 5A). Specificity of the LO1-H3 interaction was demonstrated by the virtual absence of binding of control IgG3 MAb HK-PEG-1 to any of the four constructs (Fig. 5B).

Effects of VH and VL domain swops between scFv H3 and C12: H3, C12, H3VH/C12VL, and H3VL/C12VH were coated at 10 μg/mL. Binding of MAb LO1 was tested by ELISA across a concentration range of 0.1–2000 ng/mL.
The H3 and C12 scFv differ in 15 out of the 18 amino acids, which are diverse in the Tomlinson I library (Table 1A). Since the VH of H3 was dominant for MAb LO1 binding, a series of individual point mutations were carried out on the H3VH/C12VL background, changing amino acids in the H3VH to those of C12, and amino acids in the C12VL to those of H3. Thus, this strategy looked for mutations in the H3 VH that that might reduce MAb LO1 binding and mutations in the C12VL as that might increase it. As summarized in Table 1B, MAb LO1 binding was severely reduced or completely abrogated by any change in the five H3 CDRH2 or four H3 CDRH3 amino acids to those found in C12. Although substituting the whole H3VL with that of C12 had only a minor effect (Fig. 5A), individual amino acid swops were surprisingly influential, with significant and paradoxical decreases in MAb LO1 binding caused by changing either of two amino acids in C12 CDRL2 to those on H3, but significant increases in binding of MAb LO1 to constructs with individual H3 substitutions at three of the four C12 CDRL3 sites (Table 1C). Taken together, the data strongly suggest that MAb LO1 recognizes H3 as a conformational structure rather than binding a particular linear peptide motif. This view is further supported by failed attempts by ELISA to bind MAb LO1 to linear peptides, alone or in combination, covering CDRH2 (VSDISGSNTITYA), CDRH2-FR2 interface (YADVKGRFTIS), CDRH3 (CAKDDDAFDYWG), or CDRL2 (IYYASALQSGVP) (data not shown). Furthermore, these same peptides, either alone or in combination, failed to inhibit MAb LO1 binding to H3 (data not shown).
Immunocytochemistry of atherosclerosis
Preliminary ELISA established that MAb LO1 binding was unaffected by treatment of MDA-LDL with isopropanol, which was therefore used as fixative for immunostaining arterial tissue. As shown in Figure 6, MAb LO1, but not IgG3 control, showed obvious staining of both the macrophage-rich developing atherosclerotic lesion and the media of the aortic root of hyperlipidemic Ldlr-/- mice but not normocholesterolemic WT mice, consistent with tissue accumulation of oxidized LDL in Ldlr-/- mice. Importantly, staining of Ldlr-/- mouse aortic root was inhibited by MDA-LDL or scFv H3 but not by scFv C12 (Fig. 6D–F). Taken together with the inhibition of MAb LO1 binding to MDA/Cu-oxLDL by Cu-oxLDL (Fig. 1B) or H3 (Fig. 3B), these data support the specific recognition of oxidized LDL by MAb LO1 through cognate antibody-antigen recognition rather than through non-specific binding.

Immunocytochemical staining of Ldlr-/- mouse aortic root with MAb LO1. (
When applied to human carotid endarterectomy tissue, MAb LO1 showed more restricted staining compared with staining of the Ldlr-/- mouse tissue. Thus, MAb LO1 but not IgG3 control stained intracellular deposits in macrophages, and occasional extracellular deposits adjacent to the edge of the lipid necrotic core, but medial tissue was not stained (Fig. 7). This pattern is consistent with that expected in the distribution of heavily oxidized LDL in a human atherosclerotic plaque.

Immunocytochemical staining of human carotid atherosclerotic plaque with MAb LO1. (
Discussion
In this report, we describe the characterization of MAb LO1, a novel IgG autoantibody that was selected as reacting with oxidized LDL. The approach of generating MAb from the spleens of atherosclerotic mice provides a means to dissect the naturally occurring autoantibody repertoire against modified forms of LDL. Earlier reports have highlighted the high frequency of hybridomas secreting IgM germline encoded antibodies reacting with phosphorylcholine and other oxidation-specific determinants.(28,29) Out of an interest in the IgG response, we deliberately fused splenocytes from a relatively old (i.e., one year) mouse and screened for clones secreting IgG rather than IgM antibodies. The low frequency of hybridomas secreting IgG autoantibodies against oxidized LDL is similar to the experience of Witztum and associates who failed to isolate such clones.(28) Why hybridomas making IgG anti-oxidized LDL antibodies are hard to generate is not clear, as these antibodies are readily detected in serum. Clearly, however, the antibodies found following splenocyte fusion are dependent upon the antigens used for screening, and ongoing work in our laboratory is also focused on other novel autoantibodies derived from Ldlr-/- mice with specificities distinct from MAb LO1.
The copper-oxidized LDL preparation that was used to screen our hybridomas is a complex preparation likely to contain MDA-LDL as well as other oxidative modifications.(30) While MAb LO1 was subsequently found to react well, if not better, with LDL conjugated selectively with MDA compared to crude Cu-oxLDL, there was no reactivity with other forms of modified LDL tested, such as by trypsin digestion or incubation with hypochlorite. However we have not exhaustively tested binding of MAb LO1 to LDL with other possible adducts found in Cu-oxLDL, such as with 4-hydoxynonenal,(28) and we do not therefore yet know how necessary MDA-conjugation to LDL is for MAb LO1 reactivity. We did find, however, that MAb LO1 did not bind MDA-conjugated human serum albumin, indicating that the LDL carrier is as important as the MDA hapten. Furthermore, we cannot rule out the possibility that MDA is irrelevant to binding, and that what is critical is the unmasking of a cryptic epitope on ApoB with the structural changes to the LDL particle that occur with oxidation.
Staining of atherosclerotic plaques has been used by several groups to validate the binding of antibodies against modified LDL with naturally occurring epitopes in tissues.(22–24,26,28,29) In a similar way, we have found that MAb LO1 specifically binds antigen accumulated in the aortic root of Ldlr-/- mice and in foam cells and extracellular aggregates adjacent to the necrotic core within clinical pathological carotid tissue (AHA Type IV or so-called vulnerable plaque with an advanced lipid necrotic core and thin fibrous cap). In this area, it is thought that macrophages further modify mildly modified LDL, and are killed by their load of heavily modified LDL. The selectivity of LO1 staining for this zone of the plaque is consistent with current ideas about plaque LDL handling. Most of the extracellular LO1 staining (i.e., not obviously around nuclei) was also cell shaped, reflecting either that the nucleus was out of section (in the z-plane) or reflecting release from dying cells.(41) Importantly, the isotype control did not share this pattern. Corroborating this interpretation was the colocalization of LO1 specific staining (not found in the isotype control) in the Alexafluor 568 (red emission) channel with classical oxidized LDL-associated autofluorescence in the green emission channel (not shown).(42) It was notable that the diffuse staining, including the media that was observed in the Ldlr-/- mouse tissue, was not seen in the human carotid, perhaps reflecting a more widespread microscopic accumulation of oxidized LDL in the hypercholesterolemic mouse.
It is interesting that MAb LO1 is of the IgG3 isotype, as mouse IgG3 is a relatively minor isotype in mouse serum (0.1–0.2 mg/mL, ∼2% of total IgG; compared with IgG2a/c ∼0.4 mg/mL). Mouse IgG3 activates complement well (IgG3,2a,2b> IgG1), probably as cooperative binding of its Fc domains gives IgG3 a functional polyvalency for C1q similar to IgM.(43–45) In contrast to other mouse IgG isotypes, IgG3 does not ligate proinflammatory Fcγ receptors (IgG1,2a/c,2b>> IgG3 in mice).(13,46,47) Thus, IgG3 antibodies may play a similar homeostatic role to IgM in scavenging modified LDL and, furthermore, are able to perform feedback enhancement of antibody responses.(48) By virtue of smaller monomeric size (∼150 kDa versus ∼900 kDa for IgM), IgG3 is likely to have greater tissue penetration and distribution than IgM.(49) Secretion of IgG3 is thought to be by B1 cells and to occur early in the adaptive immune response by thymus-independent (TI) class-switching with minimal hypermutations.(45,50)
Low density lipoprotein is a 18–25 nm diameter particle (Mr 550 kDa) consisting of a hydrophobic triglyceride and cholesterol ester core, surrounded by a phospholipid shell wrapped by a single apolipoprotein apoB molecule, which in humans has 4,536 amino acids.(51) The failure of isopropanol to influence the antigenicity of MDA-LDL for MAb LO1 suggests that the epitope is made up from protein rather than lipid determinants. To gain more insight into the binding interaction between MAb LO1 and the large and complex LDL particle, we screened an scFv library for an anti-idiotype antibody that might mimic some or all of the epitope on MDA-LDL by carrying an “internal image” of the antigen. It should be noted that mutating amino acids in the binding site of a MAb against apoB has previously been used to gain insight into the molecular basis of LDL interaction with the LDL receptor,(52) but an anti-idiotype approach to dissecting an anti-oxidized LDL antoantibody does not appear to have been previously adopted. We succeeded in isolating H3, a scFv antibody that specifically neutralizes the binding of MAb LO1 to MDA-LDL and which has amino acid sequences in the H3 CDRH2 and CDRL2 with strong similarity to peptide stretches in mouse and human ApoB. It is notable that while MAb LO1 showed similar physico-chemical constraints in binding H3 as MDA-LDL, the pH tolerance of H3 binding was greater, suggesting that H3 may actually be a better fit for the MAb LO1 antigen binding site than MDA-LDL. This may be related to MAb LO1 VH being germline and lacking hypermutated sequences.
Further dissection of MAb LO1-H3 interactions in comparison with a non-binding control scFv C12 showed that the MAb LO1 bound well to H3VH paired with C12VL, demonstrating the importance of VH residues. Site-directed mutagenesis failed to reveal particular H3VH amino acids critical for binding, and the major reduction in binding that occurred with any substitution suggests that CDRH2 and CDRH3 are both involved. It is notable that CDRH3 contains a stretch of six amino acids with four aspartates (DDDAFD), which may well supply a critical negative charge. It should be noted that the failure of MAb LO1 to bind to H3VL paired with C12VH does not exclude involvement of the VL, since amino acid substitutions in the two diverse amino acids at CDRL2 abolished binding. Furthermore, SALQSG, which are the amino acids in CDRL2 with homology to apoB, are conserved between H3 and C12. Thus residues on H3VL, particularly in CDRL2, may contribute to the putative conformational epitope that MAb LO1 reacts with.
Taken together, our observations raise the possibility that H3 has focal sequence and charge similarities to the site on oxidized LDL that binds MAb LO1. Linear peptides spanning H3 CDRH2, CDRH3, and CDRL2 were not able to inhibit MAb LO1 binding to H3, consistent with the importance of conformation. Therefore definitive evidence for the residues on H3 that MAb LO1 reacts with will require further studies. While there are precedents for co-crystallization of Fab with anti-idiotype,(53) this would be technically extremely difficult for MAb LO1 bound to oxidized LDL. However an alternative approach for directly comparing MAb LO1 binding to H3 and oxidized LDL is the use of hydrogen-deuterium exchange mass spectrometry.(54)
While phage display techniques have previously been used to isolate human IgG single-chain autoantibodies reacting with oxidized LDL,(20,55) as far as we are aware MAb LO1 is the first spontaneously arising IgG anti-oxidized LDL autoantibody to be reported from mice. The unique autoantibody-anti-idiotype combination provided by MAb LO1 and H3 will now be very useful for further structural and functional studies and will also allow us to perform in vivo experiments testing the capacity of passive immunization with MAb L01 to influence oxLDL clearance in vivo and to retard atherosclerosis, and also examining the possibility that active immunization with H3 can elicit an endogenous LO1-like IgG autoantibody response.
Footnotes
Acknowledgments
This work received CMRPG350214 funding from Chang-Gung Hospital (Taiwan, to S.H.C.; from the British Heart Foundation (BHF Gerry Turner Intermediate Clinical Research Fellowship FS/07/010 to J.B. and core professorial support to D.H.): and from the Higher Education Commission (Government of Pakistan, Islamabad, Pakistan, to M.Z. Dogar). The atherosclerosis staining was made possible by funding through the National Institute of Health Research Comprehensive Biomedical Research Centre at Imperial College Healthcare NHS Trust.
Author Disclosure Statement
The authors have no financial interests to disclose.