
Abstract
Antibody purification using proteins A and G has been a standard method for research and industrial processes. The conventional method, however, includes a three-step process, including buffer exchange, before chromatography. In addition, proteins A and G require low pH elution, which causes antibody aggregation and inactivates the antibody's immunity. This report proposes a two-step method using hydroxyapatite chromatography and membrane filtration, without proteins A and G. This novel method shortens the running time to one-third the conventional method for each cycle. Using our two-step method, 90.2% of the monoclonal antibodies purified were recovered in the elution fraction, the purity achieved was >90%, and most of the antigen-specific activity was retained. This report suggests that the two-step method using hydroxyapatite chromatography and membrane filtration should be considered as an alternative to purification using proteins A and G.
Introduction
HAp, which is downstream in the conventional process, has amphoteric ion-exchange properties and metal affinity because of included phosphate and calcium sites.(2) The special characteristics of HAp provide a good separation capacity.(3) Therefore, we propose that MAb could be purified by the sole use of HAp chromatography, without proteins A and G. This report describes this novel MAb purification method. In addition, a two-step purification method using HAp chromatography and filtration is also introduced for further purification.
Materials and Methods
Hybridoma preparation
Spleen cells were obtained from the inoculated mouse with Japanese encephalitis virus. The cells were fused with myeloma cells (SP2/O) in 50% polyethylene glycol/5% DMSO. The fusion cells were cultured for 2 weeks in hypoxanthine-aminopterine-thymidine (HAT) selective medium (Invitrogen, Carlsbad, CA). A hybridoma cell that produces a specific antibody was selected and cloned.
Culture medium
The culture fluid used was a serum-free medium (Hybridoma-SFM, Invitrogen) supplemented with L-glutamine (4 mM), sodium pyruvate (1 mM), and penicillin (100 U/mL)/streptomycin (0.1 mg/mL). The numbers in parentheses are the final concentrations.
Monoclonal antibody production
Anti-Japanese encephalitis virus monoclonal mouse IgG was produced by hybridoma cells. A total of 100×104 cells/mL of the hybridoma cells were suspended in 50 mL of the medium in a 75 cm2 suspension culture flask. After culturing for 14 days, the serum-free supernatant was harvested by centrifugation and filtered through a 0.22 μm membrane (Millipore, Billerica, MA) to remove the admixture substances. The monoclonal antibody was determined to be subclass IgG1 by use of the ImmunoPure Monoclonal Antibody Isotyping Kit I (Thermo Fisher Scientific, Rockford, IL).
MAb purification by HAp chromatography
Chromatography was performed with BioLogic DuoFlow (Bio-Rad, Hercules, CA). Ceramic HAp (CHT Type II, particle size 40 μm, Bio-Rad Laboratories) was packed into a stainless steel column (4.0 mm I.D.×100 mm, Sugiyama Shoji, Kanagawa, Japan). The initial buffer A was 10 mM sodium phosphate buffer (NaPB, pH 6.8). The elution buffer B was 20 mM NaPB including 1 M sodium chloride (pH 6.8). The final buffer C was 400 mM NaPB (pH 6.8). The column was washed with 8 column volumes (CV) of buffer C and equilibrated with 16 CV of buffer A. Then, 10 mL of culture supernatant as prepared in the Monoclonal Antibody production section (previous page) were directly loaded onto the HAp column, without buffer exchange. The chromatographic procedure was performed according to the following protocol: 100% buffer A (4 CV), 25% buffer B with 75% buffer A (12 CV), 100% buffer B (8 CV), and 100% buffer C (4 CV). The fractions were collected every 1 min. The flow rate was adjusted throughout, at 1 mL/min. UV absorbance of the effluents was monitored at 280 nm.
Purification by membrane filtration
The fractions from 35 to 38 min were mixed and purified by use of a PES MWCO 50,000 membrane (Sartorius Stedim Biotech, Aubagne, France) to remove low molecular admixture proteins.
Assays
The quantity of MAb in each fraction was determined by enzyme-linked immunosorbent assay (ELISA). The activity of MAb purified was also examined by ELISA. Double strand DNA (dsDNA) in each fraction was quantified by fluorescent detection assay, using Quant-iT PicoGreen dsDNA Assay kit (Invitrogen). The absorbance and fluorescence intensities were measured with a micro plate reader (Genios, Tecan, Männedorf, Switzerland).
SDS-PAGE
Fractions were analyzed by SDS-PAGE using 10% polyacrylamide separating gel (ATTO, Tokyo, Japan) without a reducing agent. After electrophoresis, the gel was visualized by use of silver staining, according to the Morrissey method.(4) The gel image was analyzed by densitometry using QScan image analysis software (Biosoft, Cambridge, United Kingdom) to evaluate the purity of the MAb in the fractions.
Results and Discussion
Comparison and suggestion of MAb purification
Figure 1 shows the scheme of the MAb purification process. In conventional methods (process A), three-step processes are ordinarily required for MAb purification using proteins A and G, followed by HAp or other ion exchange chromatography. Furthermore, complicated buffer exchange is required before chromatography. Buffer exchange procedure has been improved to high-throughput by some researchers; however, it is best to skip the buffer exchange steps.(5) In contrast, we have proposed a novel and simple MAb purification process that uses HAp chromatography, followed only by membrane filtration (Fig. 1, process B). The two-step process requires no buffer exchange; therefore, process B shortened the running time to a one-third of process A for each cycle. This novel method is clearly efficient, and the availability is described below.
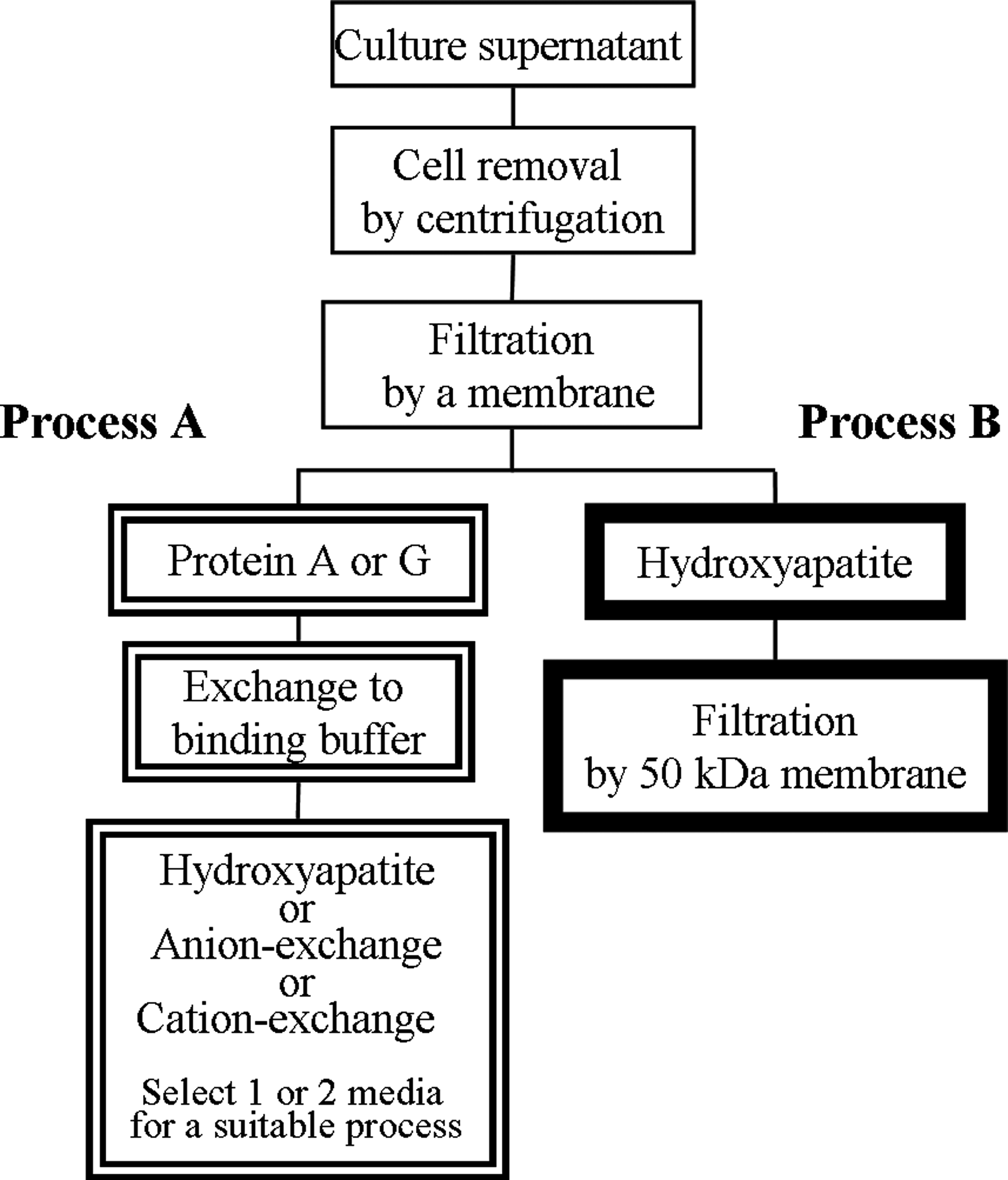
Comparison and suggestion of MAb purification process. Process A shows the conventional method using proteins A and G, followed by HAp or ion exchange chromatography. Process B shows our newly proposed method using HAp chromatography and filtration.
MAb separation by HAp chromatography
Hybridoma culture supernatant was purified by HAp chromatography, and each fraction was analyzed by ELISA and dsDNA detection (Fig. 2A). The three elution peaks appeared at retention times of 21 min (Fr. 1), 35 min (Fr. 2), and 48 min (Fr. 3), for the first, second, and third peak, respectively. As a result of ELISA, a higher quantity of MAb was detected at the Fr. 2 than at other fractions. The recovery of the MAb in Fr. 2 was 90.2% determined by ELISA results, whereas only 9.1% of MAb was distributed between Fr. 1 and Fr. 2. Therefore, almost all MAb was eluted and recovered in Fr. 2. Recovery could be improved to nearly 100%, if more sensitive elution conditions were used.

(
On the other hand, host cell-derived dsDNA in the fractions was quantified by fluorescent detection assay. Figure 2A shows that all dsDNA was eluted in Fr. 3, and dsDNA was not detected by fluorescent detection assay (detection limit 5 ng/mL) in any of the other fractions. These results indicated that nearly 100% of the host cell-derived dsDNA was removed from MAb by HAp chromatography during the purification process.
SDS-PAGE analysis
Each fraction from 35 to 38 min (represented as Fr. 2a, Fr. 2b, and Fr. 2c) was analyzed with SDS-PAGE (Fig. 2B). Mixed fractions of Fr. 2a–2c were also analyzed with SDS-PAGE, after filtration through a PES MWCO 50,000 membrane (Fig. 2B). Two major bands were observed at Fr. 2a–2c (lanes 4–6). The high-molecular-weight band of the two (asterisk 1) indicates MAb according to Western blot analysis (laboratory data). Purities of MAb were 24% (Fr. 2a), 35% (Fr. 2b), and 25% (Fr. 2c) by gel image analysis. Similarly, the low-molecular-weight band (asterisk 2) represented admixture proteins, not MAb, and the abundance ratio was 69% (Fr. 2a), 50% (Fr. 2b), and 71% (Fr. 2c). Furthermore, the 80 kDa minor protein (closed arrowhead) would be the transferrin originally included in culture medium.
On the other hand, in the mixed fraction group, the low-molecular-weight band was removed and disappeared by filtration; therefore, almost a single band could be observed, and the purity was 90% (lane 7). Purity could be improved to nearly 100%, if Coomassie Brilliant Blue staining were used as in the conventional studies.(6) We intentionally used highly sensitive silver staining for further examination, even if the purity seemed low. In addition, our two-step method had an effect on the immunity of antibody, because the antigen-specific activity of MAb showed 103 and 3791% by the purification in comparison with culture medium and commercially available polyclonal mouse IgG, respectively, from ELISA assay.
At the very least, these results indicated that the two-step purification method for MAb is available, using HAp chromatography, followed by membrane filtration, without proteins A and G. Furthermore, the two-step method will be beneficial in cases where proteins A and G cause aggregation problems and cannot be adopted for any antibodies in efficient condition.(7)
Conclusion
The conventional MAb purification process has comprised three steps using proteins A and G, followed by HAp or other ion exchange chromatography. Although MAb aggregations are frequently formed under low pH condition, proteins A and G have been widely used. In this report, we have proposed the only two-step MAb purification process using HAp chromatography and membrane filtration, without proteins A and G. This method does not require low pH elution, and can shorten the run time to one-third the conventional method for each cycle. During HAp chromatography, almost all MAb was recovered in the elution fraction while retaining the activity of antibody, and host cell-derived dsDNA was removed from the MAb. After HAp chromatography, the purity of the MAb was greater than 90% by membrane filtration. This suggests that our MAb purification method can be an alternative to the conventional processes using proteins A and G.
Footnotes
Acknowledgment
The authors thank Mr. Ken Sugo (Hoya Corporation) for his advice in the writing of this report.
Author Disclosure Statement
The authors have no financial interests to disclose.