
Abstract
Sapovirus (SaV) is an agent of human and porcine gastroenteritis and a member of the family Caliciviridae. SaV has been classified based on VP1 full gene nucleotide sequences into five genogroups (GI–GV), among which GIII is known to infect pigs. The VP1 folds into two major domains designated S and P for the shell and protruding domain, respectively. The P domain is divided into two subdomains, P1 and P2. In this study, the VP1 full gene and the S, P, and P2 regions of the VP1 gene of porcine SaV were expressed using a baculovirus expression system. Expressed proteins in the recombinant virus were confirmed by polymerase chain reaction, indirect fluorescence antibody (IFA) testing, and Western blot analysis. Four hybridomas secreting VP1-specific monoclonal antibodies (MAbs) against porcine sapovirus were generated. Four MAbs were characterized according to their IFA and Western blot analysis results. All of the hybridomas produced in this study secreted MAbs binding to S domain of VP1 protein specifically. The MAbs produced in this study can be used as specific diagnostic reagents for detecting porcine SaV.
Introduction
SaVs were discovered for the first time by electron microscopy (EM) in fecal samples from infants suffering from a gastroenteritis outbreak in an orphanage in Sapporo, Japan, in 1977.(7) For this reason, the virus was initially known as Sapporo virus (SV). Subsequently, the virus has been renamed SaV in 2002. In 1990, porcine SaVs were first identified in US piglet fecal samples by EM(8) and classified genetically as SaV in 1999.(9) Subsequently, porcine SaVs have been reported in Europe, Asia, and the United States, suggesting widespread distribution.(10–12) SaVs have been identified in both diarrheic and asymptomatic pigs of different ages.(11) Most porcine SaV strains detected so far have been classified as Cowden-like G III SaVs.(11,12) However, a number of novel and variant animal SaV strains distantly related to typical swine GIII and human strains have been detected in mink and swine.(13)
The SaV genome, which is polyadenylated at its 3′ terminus, is a linear, single-stranded, positive-sense RNA molecule of approximately 7.5 kb with either two or three open reading frames (ORFs).(14) ORF I encodes a 250 kb polyprotein that includes amino acid motifs characteristic of caliciviruses, containing 2C-like NTPase, VPg, 3C-like protease, 3D-like RNA-dependent RNA polymerase, and capsid protein (VP1). ORFs I and II encode a small, basic protein but the function of the protein is unknown. Based on a phylogenetic analysis of the VP1 full-gene sequence, SaVs have been divided into five genogroups (GI–GV).(6) SaV GI and GII have three genotypes, respectively, whereas SaV GIII, GIV, and GV have one genotype. SaV GI, GIV, and GV possess three ORFs, whereas SaV GII and GIII contain only two ORFs. Human SaVs belong to GI, GII, GIV, and GV, whereas porcine SaVs belong to GIII. Recently, new porcine SaVs genogroup (GVI, GVII, and GVIII) have been proposed.(10,13) Most porcine SaVs that belong to GIII SaVs cannot be grown in cell culture, but the Cowden strain was adapted to grow in a continuous cell line(15); however, intestinal fluid filtered from uninfected gnotobiotic pigs is needed in the virus growth medium for cell culture. Chang and associates reported that bile acids supply important factors to support virus replication.(16)
VP1, which is the major SaV capsid protein, is responsible for most capsid-related functions such as assembly, host interaction, and immunogenicity.(17) VP1 is comprised of a shell domain (S) linked by a flexible hinge to a protruding (P) domain. The S domain forms the inner part of the capsid, which surrounds the RNA genome and maintains the icosahedral structure, and the P domain forms the arch-like protrusions that emanate from the shell and contain dimeric contacts.(18) The P domain is further divided into two subdomains, P1 and P2. The P1 subdomain forms the sides of the capsomere arch, and the P2 subdomain is positioned at the top of the arch. The exposed VP1 variable region on the surface of the virion coincides with the suspected role of VP1, which is the formation of the major antigenic site and the interaction between the receptor and host cells.(18–20) The highly conserved S domain may be responsible for the icosahedral scaffold, offering an N-terminal arm to promote the appropriate curvature as a switch. The hypervariable P domain may function as a replaceable module to produce strain differences and antigen specificity.(17,18) Direct support for this hypothesis has been found in recent studies that show the P2 domain as an important site conferring antigenic and receptor binding specificity of NoVs.(20,21)
As stated above, SaVs are uncultivable with the exception of the Cowden strain. Therefore, SaVs were originally detected using EM, but this method is unsuitable for examining large numbers of specimens, and the viruses are difficult to identify due to their small size. Currently, reverse transcription-polymerase chain reaction (RT-PCR), which has high sensitivity and can be used for a genetics analysis, has been widely used as a diagnostic tool to detect SaVs.(22) Several groups have purified native SaV particles and produced antisera against them for use in enzyme-linked immunosorbent assays (ELISAs) and other immunoassays.(23–25) However, no diagnostic tests for porcine SaVs have been developed for use outside the research laboratory. An antigen ELISA was developed and used to study porcine enteric calicivirus.(26)
In this study, the VP1 full gene and the S, P, and P2 regions of the porcine SaV VP1 gene, which are useful as diagnostic reagents for an ELISA, were expressed using a baculovirus expression system. We also generated and characterized monoclonal antibodies (MAbs) against VP1.
Materials and Methods
Viruses and RNA extraction
The plasmid containing the VP1 gene of porcine SaV Cowden strain used in this study was provided by Professor Cho (Chonnam National University, Gwangju, Korea). SaV DNA was extracted using the Viral Gene-spin Viral DNA/RNA Extraction kit (Intron, Seongnam Si, Korea). Four primer sets based on the conserved sequences of the SaV VP1 gene, and the P, S, and P2 domains were prepared to amplify them individually in porcine SaV (Table 1).
Enzyme site: XhoI, CTCGAG; HindIII, AAGCTT.
For virus cross-reactivity test, recombinant VP1 protein of porcine NoV, bovine NoV, and human NoV were used. Three recombinant proteins were confirmed as norovirus by RT-PCR from fecal sample of pig, cow, and human, respectively. Three recombinant proteins were confirmed as norovirus by RT-PCR from fecal sample of pig, cow, and human, respectively; their VP1 genes were expressed in baculovirus expression vector system.
Cloning of VP1 gene of SaV
Each PCR product was digested with the Xho I and Hind III enzymes and inserted into the XhoI and Hind III sites of the pBlueBac4.5/V5-His-TOPOR vector (Invitrogen, Carlsbad, CA) using T4 DNA ligase (Takara, Tokyo, Japan). The ligation mixtures were then transformed into competent E. coli DH5α (Invitrogen). The recombinant vectors were collected and checked for the insertion by DNA sequencing.
VP1 gene expression
Recombinant baculoviruses were generated by co-transfecting insect cells with Bac-N-Blue DNA (Invitrogen) and a baculovirus transfer vector containing the VP1 gene (P, S, and P2 domains). The recombinant baculovirus production procedure was performed according to the Bac-N-Blue Transfection and Expression Guide (Invitrogen). Each recombinant baculovirus was plaque purified using the following procedures. Recombinant protein was purified under the native condition according to the manual provided with the QIAexpress Ni-NTA Fast Start kit (Qiagen, Hilden, Germany).
Immunization of mice
BALB/c mice (4–5-week-old male) were immunized, and the VP1 recombinant protein was purified to produce VP1-specific MAbs. Briefly, purified VP1 recombinant protein was emulsified in an equal volume of Freund's adjuvant and then inoculated into each footpad of the BALB/c mice. The inoculation was repeated every 3 days. Purified protein was emulsified in complete Freund's adjuvant for the first inoculation, whereas incomplete Freund's adjuvant was used for the two subsequent inoculations.
Production of MAbs
Cells from spleen and popliteal lymph nodes of mice were fused with Sp2/O myeloma cells following the procedure of Kang and associates, with minor modifications.(27) Hybridoma culture supernatants were screened for MAb production against porcine SaV using an indirect fluorescent antibody (IFA) test. Hybridomas that were positive against porcine SaV in the IFA test were cloned at least twice by limiting dilution techniques. Ascites was prepared by injecting hybridoma cells into pristane-primed mice. Ascites were harvested at 8–10 days post-injection and stored frozen.
MAb characterization
The isotype of each MAb was determined using a MAb Isotyping kit (Sigma, Korea) according to the manufacturer's instructions. MAb domain specificity was determined by IFA. IFA was used to determine the cross-reactivities of the MAbs with other recombinant NoVs.
Indirect fluorescent antibody
Recombinant baculovirus was inoculated into the Sf9 cell monolayer prepared in a 96-well plate. When cytopathic effects (CPE) appeared, the cells were fixed with 80% acetone for 7 min, the plate was washed three times with PBS (pH 7.2) and then air dried. The fixed cells were reacted with 100 μL of undiluted hybridoma supernatant or diluted ascites and incubated at 37°C for 1 h. After washing three times with PBS, the cells were stained with 50 μL of diluted FITC-conjugated goat anti-mouse IgG+M (ImmunoResearch Lab, West Grove, PA) and incubated at 37°C for 1 h. Finally, the cells were washed three times with PBS, mounted in buffered glycerol (80%), and examined under fluorescence microscopy (Olympus, Tokyo, Japan).
Western blot analysis
Recombinant baculovirus was inoculated into the Sf9 cell monolayer. When CPE appeared, cells infected with recombinant baculovirus were collected and centrifuged for 20 min at 2,000 rpm, and the supernatant was removed. The pelleted material was resuspended individually in 1 mL PBS, extracted three times through freeze/thawing, and then lysed by sonication. Western blotting was performed according to methods previously described with minor modifications.(28) Briefly, the recombinant proteins were electrophoresed on 12% vertical SDS-PAGE and transferred to nitrocellulose (NC) membranes. The NC membrane was blocked with 5% skim milk in TBS-T at 37°C for 2 h. The membrane was reacted with supernatant from the hybridoma culture or ascites for 1 h at room temperature (RT) with shaking. After washing with TBS-T, diluted alkaline phosphate-conjugated goat anti-mouse IgG+M was added and incubated for 1 h at RT. After washing the membrane with TBS-T, BCIP/NBT (Promega, Madison, WI) was added as a substrate and incubated until a band appeared. The reaction was stopped by washing with tap water.
Results
VP1 gene cloning
Amplification of the VP1, P, S, and P2 domains of porcine SaV (Cowden) was achieved using PCR with specific primer sets. The amplified VP1 gene and the P, S, and P2 domains were 1626 bp, 984 bp, 642 bp, and 456 bp in size, respectively (Fig. 1).
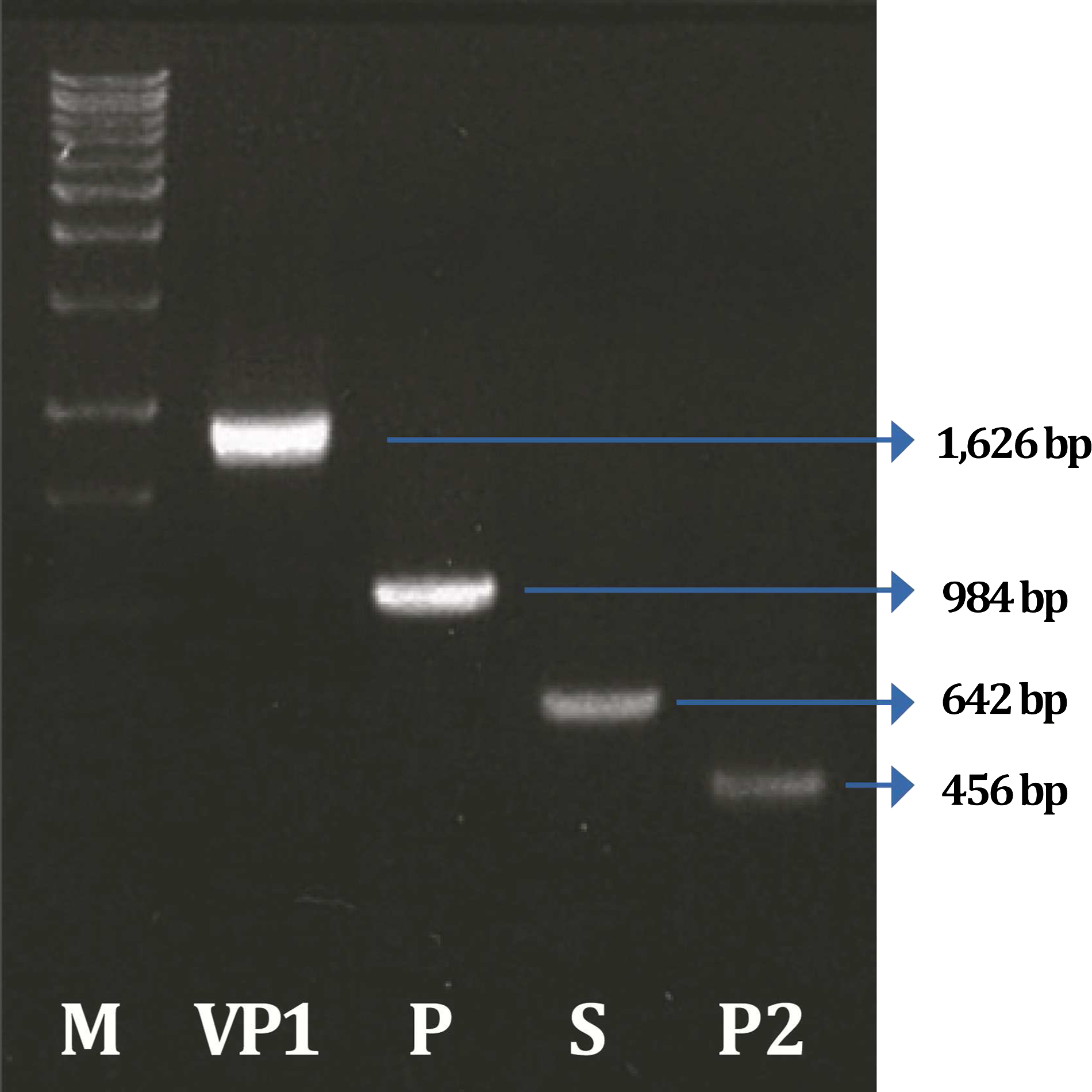
Amplification of porcine SaV VP1 gene (P, S, and P2 domains) by PCR.
VP1 gene expression
Recombinant baculovirus was generated by co-transfecting insect cells with Bac-N-Blue DNA and the pBlueBac4.5/V5-His-TOPO transfer vector containing the VP1 gene, and the P, S, and P2 domains. Recombinant baculoviruses were confirmed to determine wild-type contamination by PCR using specific primers for VP1, P, S, P2, and the baculovirus polyhedrin gene. As a result of PCR using gene-specific primers, the amplified VP1 gene (1626 bp) and the P (984 bp), S (642 bp), and P2 (456 bp) domains were in agreement with expected sizes. As a result of PCR using baculovirus polyhedrin gene-specific primers, the amplified VP1 (2167 bp), P (1525 bp), S (1183 bp), and P2 (997 bp), containing DNA (541 bp) contributed by the transfer vector, were in agreement with expected sizes (Fig. 2).
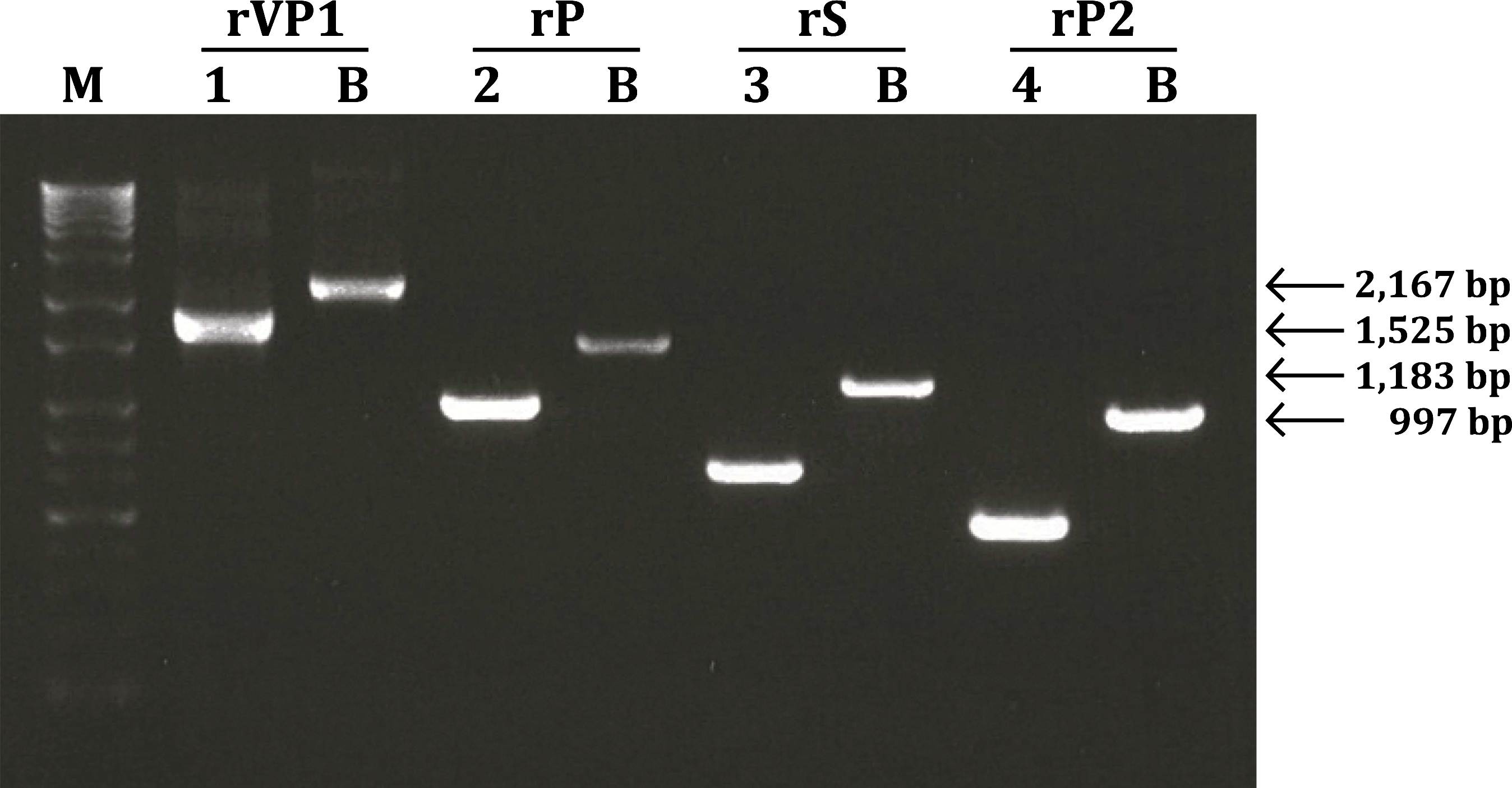
Confirmation of porcine SaV VP1 gene (P, S, and P2 domains) recombinant baculovirus by PCR. 1–4, porcine SaV gene-specific primers; B, baculovirus polyhedrin gene-specific primers. M, 1Kb+ladder marker.
The cells were infected with each recombinant baculovirus to confirm recombinant protein expression. As a result, CPEs were not observed in mock-infected Sf9 cells. In contrast, a baculovirus-specific CPE such as swelling was observed in Sf9 cells infected with each recombinant baculovirus. Recombinant protein expressed in the baculovirus expression system was confirmed for antigenicity using IFA and Western blotting with the anti-V5 specific antibody. The IFA analysis showed strong fluorescence in Sf9 cells inoculated with each of the recombinant baculoviruses. However, no fluorescence was detected in the control group (Fig. 3). Western blot analysis showed that the 60.5 kDa (VP1), 37 kDa (P), 24 kDa (S), and 17 kDa (P2) sizes for the protein bands were in agreement with the expected molecular mass of the protein identified from Sf9 cell lysates infected with each recombinant virus. The P domain was expressed at high levels, similar to those exhibited by the VP1 recombinant protein, whereas the S and P2 domains were expressed at relatively lower levels (Fig. 4).
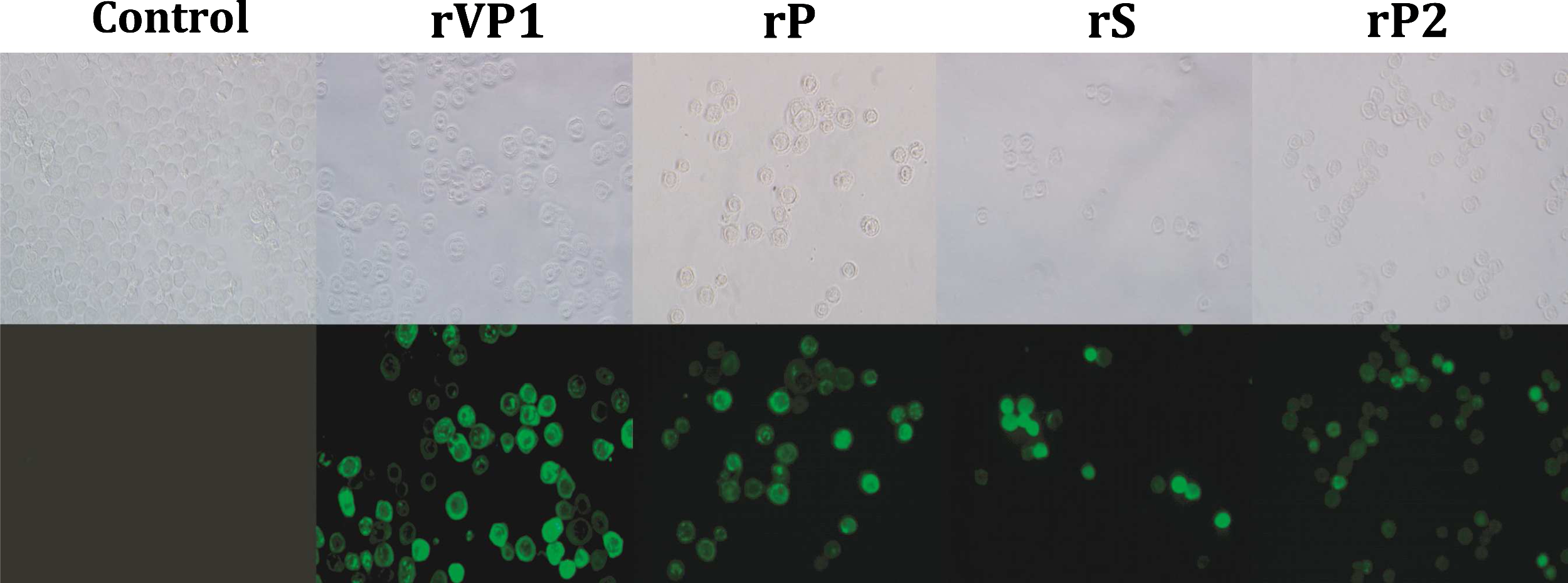
Cytopathic effects and immunofluorescences of Sf9 cells infected with porcine SaV VP1 gene (P, S, and P2 domains) recombinant baculovirus. Control, mock-infected Sf9 cells; rVP1, porcine SaV VP1 recombinant baculovirus; rP, porcine SaV P recombinant baculovirus; rS, porcine SaV S recombinant baculovirus; rP2, porcine SaV P2 recombinant baculovirus.

Western blot analysis of expressed porcine SaV VP1, P, S, and P2 recombinant proteins using V5-specific antibody. 1, porcine SaV VP1 recombinant baculovirus; 2, porcine SaV P recombinant baculovirus; 3, porcine SaV S recombinant baculovirus; 4, porcine SaV P2 recombinant baculovirus; 5, mock-infected Sf9 cell lysate; M, molecular weight marker.
Characterization of VP1 MAbs
A total of four MAbs were produced. The characteristics of the four MAbs against porcine SaV VP1 recombinant proteins expressed in the baculovirus expression system are shown in Table 2. The isotype of MAb 6E5 was IgG2b. The isotype of MAbs 6H3, 5B3, and 3B10 was IgM. Antigenicity and domain specificity of the MAbs were confirmed by IFA and Western blotting using cells infected with each recombinant baculovirus (rVP1, rP, rS, and rP2). IFA analysis showed that all recombinant proteins expressed by rVP1, rP, rS, and rP2 reacted strongly with the anti-V5 specific antibody, whereas all of the MAbs only reacted with recombinant VP1 and S protein and produced strong fluorescence (Fig. 5). The MAbs also were confirmed to have a reaction with the cells infected with Sapovirus Cowden strain (data not shown). Western blot analysis showed that all of the MAbs reacted with the cell lysates, which were only infected with the S recombinant baculovirus and formed a band. However, cells lysates that were infected with VP1 recombinant baculovirus did not react with the MAbs (Fig. 6). The IFA results showed that the MAbs did not cross-react with other recombinant NoVs.
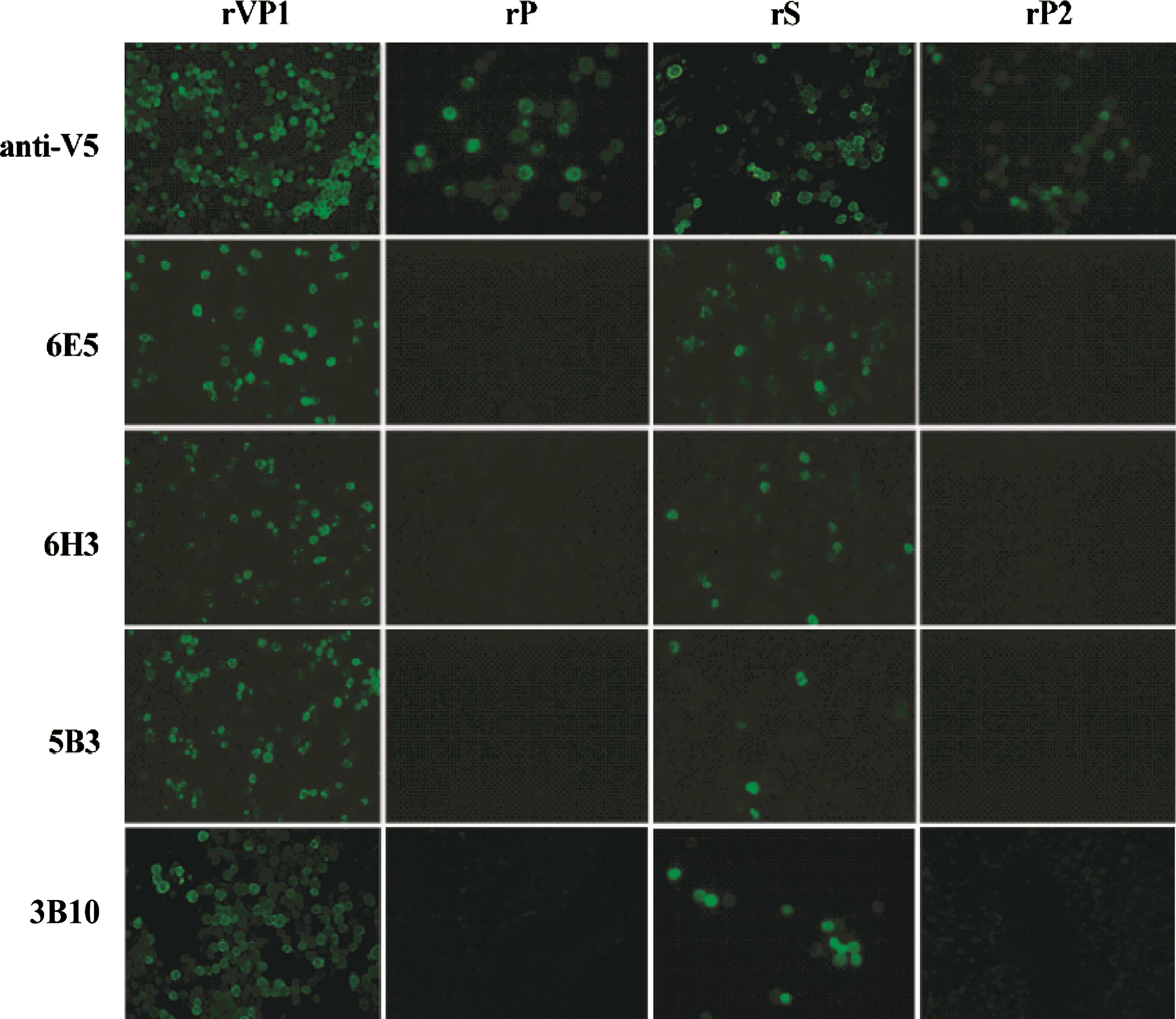
Immunofluorescence of porcine SaV VP1-specific monoclonal antibodies (MAbs). Porcine SaV VP1, P, S, and P2 recombinant baculovirus-infected Sf9 cells were reacted with MAb (6E5, 6H3, 5B3, and 3B10) and anti-V5-specific antibody. rVP1, porcine SaV VP1 recombinant baculovirus; rP, porcine SaV P recombinant baculovirus; rS, porcine SaV S recombinant baculovirus; rP2, porcine SaV P2 recombinant baculovirus.

Antigenicity of porcine SaV VP1-specific monoclonal antibodies (MAbs). 1: porcine SaV VP1 recombinant baculovirus-infected cell lysates; 2, porcine SaV P recombinant baculovirus-infected cell lysates; 3, porcine SaV S recombinant baculovirus-infected cell lysates; 4, porcine SaV P2 recombinant baculovirus-infected cell lysates; 5, mock-infected Sf9 cell lysate; M, molecular weight marker.
Reactivity with recombinant viruses (rVP1, rP, rS, and rP2) and other NoVs were confirmed by IFA.
rVP1, porcine SaV VP1 recombinant baculovirus; rP, porcine SaV P recombinant baculovirus; rS, porcine SaV S recombinant baculovirus; rP2, porcine SaV P2 recombinant baculovirus; rPorcine NoV, porcine NoV recombinant baculovirus; rBovine NoV, bovine NoV recombinant baculovirus; rHuman NoV, human NoV recombinant baculovirus.
Discussion
SaVs are enteric pathogens that cause acute gastroenteritis in humans and pigs.(29) They are distributed worldwide, and their prevalence in pig herds may be underestimated.(13) The role of the porcine SaVs between humans and animals remains uncertain, and information about the distribution and genetic diversity of porcine SaVs is still lacking.(30) The absence of clinical symptoms in animals infected with the virus indicates a possible risk to public health and sanitary conditions. Additionally, an asymptomatic infection of SaV means that the virus is circulating persistently in the pig population.(12,29) Diagnostic and epidemiological studies of human SaVs have been reported frequently, because these viruses cause gastroenteritis outbreaks in humans. In contrast, porcine SaVs, which cause diarrhea outbreaks in pigs, have not been investigated because sensitive and accredited assays for their routine detection are not available.(29)
In this study, recombinant proteins of the full VP1 gene and the S, P, and P2 regions of the porcine SaV VP1 gene, which can be used as diagnostic reagents for immunoassays, were expressed using a baculovirus expression system. In addition, MAbs against VP1 were generated and characterized.
The baculovirus expression system has been used to express many DNA and RNA viral proteins, including SaV. The baculovirus expression system has many advantages compared to other expression systems.(31–33) It has proven very effective for producing recombinant capsid proteins of NoVs and lagoviruses.(32,33) We cloned and expressed the porcine SaV VP1 gene using the baculovirus expression system. Three additional recombinant baculoviruses, which expressed the porcine SaV P, S, and P2 domains, respectively, were also cloned and expressed to determine the domain specificity of the MAbs. IFA and Western blot analyses were performed to verify recombinant protein expression. As a result, strong fluorescence was confirmed in the recombinant baculovirus infected cells, and the expected size of the protein bands was identified from cell lysates infected with each of the recombinant baculoviruses. Given these results, recombinant proteins were successfully expressed in the baculovirus expression system.
Four MAbs were produced using the porcine SaV VP1 recombinant protein, and the MAb isotypes were determined using a commercial isotyping kit. As a result, 6E5 was identified as an IgG2b isotype and 6H3, 5B3, and 3B10 were identified as the IgM isotype. Cell lysates prepared from Sf9 cell culture infected with the recombinant baculovirus expressing the different VP1 protein domains were assayed by IFA and Western blot using the MAbs produced in this study. All MAbs reacted strongly with the S recombinant protein in IFA and Western blot. However, the VP1 recombinant protein reacted strongly with all MAbs in the IFA but not in the Western blot. This result indicates that all MAbs recognized continuous epitope of the S domain of porcine SaV VP1. In contrast, the discontinuous epitope of the whole VP1 proteins were not recognized.
Several groups have reported MAbs that are specific to different VP1 protein domains and react against caliciviruses.(19,34–36) Some MAbs recognize conformational epitopes on either the N-terminus or the C-terminus,(19) whereas others react against linear epitopes on the C-terminus. Yoda and colleagues suggested that the N-terminal region of the capsid protein is more immunodominant.(36) However, other researchers have indicated that the C-terminus of the capsid protein contains the more immunodominant epitope.(19) Broadly reactive MAbs could be used as reagents for developing a simple rapid diagnostic assay for detecting SaVs, including new previously unknown SaV strains. Additionally, if SaV-specific MAbs have common epitopes among all SaVs or at least one or more genogroups of SaV, the development of a broadly reactive ELISA for detecting viral antigens would be possible. In this study, the four MAbs did not cross-react with other noroviruses. However, cross-reactivity with another SaV could not be confirmed, as most SaVs are uncultivable, so it was difficult to obtain other viruses. Additionally, due to the absence of recombinant viruses expressed by other SaVs, cross-reactivity could not be confirmed. An additional experiment will be necessary to confirm cross-reactivity with other genogroup SaVs. MAbs produced in this study were all S domain specific. We were expecting to produce various types of domain-specific monoclonal antibodies in this study. However, the results were not as expected. Although we repeated the experiment several times, only the S domain-specific monoclonal antibodies were produced. This suggests that the S domain is more immunodominant than the other domains. Also S domain is a more common epitope than the other domains. However, the number and type of MAb is too small to compare the characteristics between the domains. Therefore, further experiments are needed to produce P1 and P2 domain-specific MAbs.
The MAbs developed in this study will be very useful for developing new diagnostic tests for detecting and discriminating a wide variety of strains(35) by immunoassay such as ELISA.
Footnotes
Author Disclosure Statement
The authors have no financial interests to disclose.