
Abstract
The UL16-binding proteins (ULBPs) are a novel family of human MHC class I-related, cell surface proteins that function as ligands for NKG2D. In this study, the gene encoding human ULBP3 was cloned into prokaryotic expression vector pQE30, resulting in a recombinant plasmid pQE30-ULBP3. The pQE30-ULBP3 was transformed into Escherichia coli M15 and induced the expression of recombinant protein ULBP3 (rec-ULBP3). The purified rec-ULBP3 as an antigen was used to immunize BALB/c mice. Through cell fusion, sub-cloning, and screening approach, three hybridoma cell clones expressing monoclonal antibodies (MAb) were acquired. The results from Western blot analysis, flow cytometry, and enzyme-linked immunosorbent assay showed that the hybridoma clones B2-F1-F1 and B4-C5-D11, and not G2-A4-A12, reacted with rec-ULBP3 and nature ULBP3 expressed on the cell surface of the tumor cells. In conclusion, the new MAb described here provides a valuable tool for further investigating ULBP3 function and clinical application.
Introduction
By engaging NKG2D-activating receptor, ULBP3 can weakly activate NK cells to proliferate and secrete cytokines by triggering calcium mobilization and activation of the MEK/ERK, PI3K/Akt, and JAK2/STAT5 signaling pathways.(2) Co-stimulation of NK cells with ULBP3 and IL-12, IL-10, or IFN-α greatly enhances the production of multiple cytokines and chemokines.(2) Whether or not ULBP3 can directly activate CD8+ αβ T cells and/or γδ T cells to produce a cytotoxic effect,(3–14) it is able to enhance the antigen-dependent activation of CD8+ αβ T cells by engaging NKG2D. In vivo ULBP3 expression correlates with improved survival in chronic lymphocytic leukemia of B-cell type patients.(15) However, accumulating data suggest that the NKG2D pathway is correlated with the mechanism of immune escape in tumor immunity. It has been reported that a tumor can evade an NKG2D-mediated antitumor response by shedding its ligands from the cell surface and then down-regulating the expression of NKG2D.(16–24)
Recent studies indicate that ULBP mRNA is detected in a wide variety of tumors and cell lines, but it is undetectable in several normal tissues.(25) In this study, we produced a monoclonal antibody against ULBP3 using a human sequence-based recombinant ULBP3 protein (rec-ULBP3) as antigen. Since ULBP3 polyclonal antibodies have been used up until now, obtaining the hybridoma cell-derived monoclonal antibody against ULBP3 is an advantage for monitoring the behavior of endogenous ULBP3 in cells and investigating the functions of ULBP3.
Materials and Methods
Animals and cell lines
Six-week-old female BALB/c mice were purchased from the Center for Experimental Animals of Yangzhou University (Yangzhou, China). The mice were bred and maintained under conventional conditions. SW620 cell line was purchased from the Shanghai Cell Biology Institutes, Chinese Academy of Sciences (Shanghai, China). Fresh tumor tissues were isolated from resected tumor specimens of patients with colorectal cancer with informed consent. The study protocol was approved by the Ethics Committee of the Affiliated Hospital of Jiangsu University.
Molecular cloning of ULBP3
The following PCR primers: ULBP3-forward 5′-ATTCTTC CGTACCTGCTATT-3′ and reverse 5′-GCTATCCTTCTCCC ACTTCT-3′ were used for cloning ULBP3 (aa15-177); SW620 total RNAs were isolated using TRIzol reagent according to instruction. Oligo dT-primers were used for reverse transcription with 1 μg of total RNA in a 10 μL reaction volume (Toyobo Bio-Technology, Osaka, Japan). cDNA obtained by reverse transcription of mRNA was used as templates for polymerase chain reaction (PCR). The PCR profile was composed of 95°C for 4 min, 25 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 40 s. There was a final extension of 72°C for 10 min. The PCR product was purified with a DNA purification kit (KeyGen Biotech, Nanjing, China). After purification, the products were cloned into pMD®18-T Simple Vector (TaKaRa, Otsu, Japan). Positive transformed DH5a cell clones were confirmed by PCR and enzyme digestion of the recombinant plasmids. Those clones containing different sizes of insertion fragments were further analyzed by DNA sequencing. The expected sizes of PCR products were 489 bp for ULBP3.
Construction of recombinant plasmid encoding ULBP3
pMD18-T-ULBP3 was used as a PCR template. Forward primer 5′-
Expression of ULBP3
After the pQE30-ULBP3 was transformed into host cells E. coli M15, the expression of the protein of interest was induced using 1 mM isopropyl-β-D-thiogalactoside (IPTG) at 30°C. The induced bacteria were centrifuged at 4°C, 10,000 rpm for 2 min. The pellets were treated one time with SDS loading buffer and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins in the gels were stained with Coomassie brilliant blue dyes. Renaturation of the fusion protein ULBP3 by dialysis were performed as described(26) and purification of the protein was performed using Profinity IMAC Ni-charged resins (Bio-Rad, Hercules, CA). The protein of interest was named rec-ULBP3.
Immunization procedure
The preparation of the anti-ULBP3 antibody was performed according to previous reports with modifications. Briefly, a mouse was immunized with 1 mL purified protein (100 μg/mL) emulsified with equal amount of Freund's complete adjuvant via subcutaneous injection. The immunization was boosted twice by inoculation of the same antigen (1 mL) mixed with an equal volume of Freund's incomplete adjuvant at a three-week interval. Two weeks and 3 days before cell fusion, mice were respectively boosted by tail vein injection with 50 μg of purified rec-ULBP3 protein diluted in 300 μL of sterile PBS. One week after each booster injection, mice were tail-bled and antisera were tested for titer of anti-ULBP3 antibody by indirect ELISA.
Cell fusion and monoclonal antibody production
The spleen was removed from the spleen-immunized mouse for cell fusion. After collecting and counting, 108 spleen cells were diluted with RPMI-1640 and mixed with 107 myeloma cells (SP2/0) prepared ahead. The mixture was pelleted by centrifugation at 1000 rpm for 8 min and suspended in 0.5 mL of HAT medium (Sigma-Aldrich, St. Louis, MO). 1 mL 50%(v/v) warm PEG-2000 (37°C) was then added into a 37°C water bath as the fusing reagent to the cell mixture for 1 min of gentle stirring followed by drawing the cell mixture into the pipette. After holding still for 1.5 min, the mixture was added to a 50 mL tube and 50 mL HAT medium was then added slowly to stop the fusion. Finally the cell mixture was diluted to about 106 cells/mL with HAT medium and distributed into each well (0.1 mL/well) of 96-well culture plates to which normal mouse macrophage cells had been added ahead of time as the feeder (0.1 mL/well). Medium in wells was replaced by fresh HAT medium every other day after the cell fusion. Ten days later, HT medium (Sigma-Aldrich) was added to the wells instead of HAT medium. Positive wells were screened by detecting the culture supernatant. Then single clones of positive hybridoma cells were obtained with a limiting dilution technique and expanded stepwise. The positive hybridoma cells were injected into the intraperitoneal cavity of BALB/c mice primed in advance with liquid paraffin for antibody production. The immunoglobulin isotypes of selected MAbs clones were determined using Immuno-type mouse MAb isotyping kit (BD Biosciences, San Jose, CA) according to the manufacturer's instruction.
Titration of the antibody using ELISA
The reaction between the ULBP3 and the antibody was analyzed by indirect ELISA. Briefly, ELISA plates were coated with 100 mL renatured protein (5 μg) in carbonate bicarbonate buffer (15 mM Na2CO3, 35 mM NaHCO3 [pH 9.6]) at 4°C overnight. The next day the plates were blocked with 10% calf serum in PBS-0.05% Tween-20 (PBST) at 37°C for 2 h. Subsequently, the antigen-containing wells were incubated with serially diluted antibody or control serum from a non-immunized mouse at 37°C for 1 h, after complete washing with PBST. The plates were incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (1:5000 diluted in PBST; Beyotime, Shanghai, China) at 37°C for 1 h after three washes with PBST. The wells were incubated with o-phenylenediamine dihydrochloride substrate for 15 min after three washes with PBST. The OD450 value was determined using an ELISA plate reader after stop with stopping solution.
Western blot analysis
The bacteria bearing pQE30-ULBP3 or empty vector were induced by IPTG and the bacterial lysates were subjected to 15% SDS-PAGE. The proteins were electronically transferred to a PVDF membrane. The PVDF membrane was blocked for 1 h at 25°C using 5% non-fat dry milk in TBST followed by incubation with the anti-His (1:1000 dilution in TBST) overnight at 4°C. The membrane was then incubated with HRP-conjugated goat anti-mouse IgG (1:5000 dilution with 5% non-fat dry milk in TBST) at 37°C for 1 h. The protein bands were visualized using ECL substrate.
Flow cytometric analysis
Approximately 105 SW620 cells were incubated with MAbs (the supernatant of hybridoma clones) together for 30 min at 4°C in 1640 medium. After the cells had been washed twice with the PBS, FITC-conjugated goat anti-mouse IgG antibody (Beyotime, Shanghai, China) was added, and the cells were incubated for an additional 30 min at 4°C. Cells were analyzed after washing twice on a FACScan II flow cytometer (BD Biosciences).
Immunohistochemistry
In brief paraffin-embedded colorectal cancer and adjacent normal tissue sections were cut 4 μm thick and mounted on glass slides. The specimens were deparaffinized in toluene, rehydrated in a graded series of ethanol solutions, and incubated with 0.3% hydrogen peroxide for 30 min. After blocking with normal serum corresponding to the origin of the secondary antibodies for 30 min, the slides were incubated overnight at 4°C with primary antibodies against ULBP3. The specimens were then washed with PBS, and incubated with biotinylated secondary antibodies for 30 min at room temperature, followed by peroxidase-conjugated avidin-biotin complex and 3, 3′-diaminobenzidine (Dako, Carpinteria, CA) as chromogen. Finally, the sections were counterstained with hematoxylin.
Results
Expression and purification of recombinant ULBP3
First, a truncated gene encoding the ULBP3 gene from SW620 cell line was amplified by RT-PCR, resulting in a gene fragment of 456 bp with the deletion of the N terminal signal peptide sequence (Fig. 1A). The PCR product was inserted into Bam HI and Hind III sites of pQE30 vector and the resulting recombinant plasmid pQE30-ULBP3 was sequenced (Fig. 1B). The sequencing report showed that the gene sequence was identical to that of the published sequence (NCBI reference sequence: NM_024518.1), confirming the authenticity of the inserted heterologous gene.
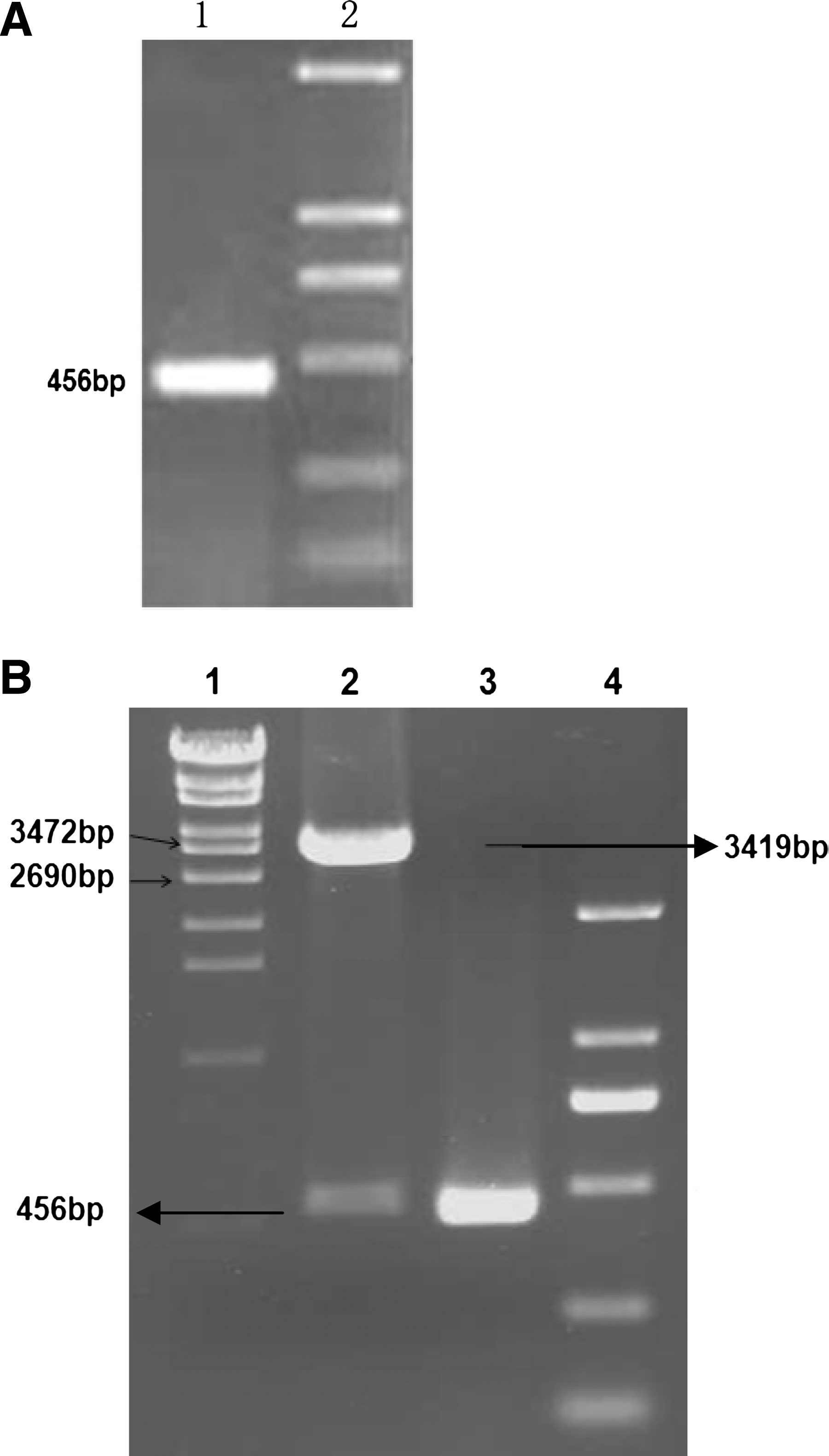
(
Second, the plasmid pQE30-ULBP3 was transformed into host bacteria M15 for expression of the recombinant protein. Our results showed that the high-level expression of rec-ULBP3 in the form of inclusion body was achieved at 4 h post-induction. In order to get a purified protein with biological activity, in this study, we utilized the fused Histidine-tag to purify and renature the rec-ULBP3 using Profinity IMAC Ni-charged resins. As shown in Figure 2A, the amount of harvested protein with a high purity was comparable to that of pre-purification. Western blot analysis showed that the anti-His antibody reacted with the rec-ULBP3 rather than the empty vector control bacteria (Fig. 2B).

(
Establishment of hybridoma cell line secreting anti-ULBP3 antibody
The purified rec-ULBP3 was renatured and used to immunize BALB/c mice. After the immunization procedure was over, ELISA results indicated that all of the titer of antisera were exceeded over 1:105. We obtained the hybridoma cell line by fusing the immune spleen cells with myeloma cell lines (SP2/0). After 2 weeks of culturing, it was found that hybridoma cell clones formed in 152 wells out of a total of 216 wells seeded with fusion cell suspension, and the ratio of fusion was about 70%. Out of 152 wells, 10 hybridoma clones were initially selected on the basis of their strong ELISA reactivities with rec-ULBP3 protein and not binding to control antigens. Through limiting dilution for subcloning and screening approach, three clones of MAb-producing hybridoma cell lines with the highest titer and good cell growth status were acquired and named as clones B2-F1-F1, B4-C5-D11, and G2-A4-A12, respectively. The three hybridoma cell clones were injected into the abdominal cavity of mice and the ascites containing MAb were acquired, respectively.
Characterization of monoclonal antibodies
Purification of the ascites was performed by protein G affinity chromatography to 90% homogeneity as assessed by SDS-PAGE. The immunoglobulin isotype analysis revealed that the subclasses of MAbs were IgG1 and IgG2a: clone B2-F1-F1 (IgG1), clone B4-C5-D11 (IgG2a), and clone G2-A4-A12 (IgG1). The titers of these purified MAb reached 5×104 – 5×105. These MAbs specifically reacted with rec-ULBP3 protein, but did not cross-react with other non-specific proteins and lysates of E. coli. To further determine the specificity of these produced MAbs, we examined whether three types of MAbs can recognize nature ULBP3 expressed on the cell surface. Flow cytometry results showed that MAbs from clones B2-F1-F1 and B4-C5-D11 reacted with the membrane ULBP3 on SW620 cells (Fig. 3A), and the extent of fluorescence signal from clone B4-C5-D11 was similar to that of clone B2-F1-F1. However, we found that MAb from clone G2-A4-A12 failed to react with the membrane ULBP3 on SW620 cells. More importantly, our findings also showed that MAbs from clone B4-C5-D11 or B2-F1-F1 reacted with the membrane ULBP3 on the primary tumor cells from colorectal cancer tissues (Fig. 3B). The result from Western blot analysis revealed that the MAb from clone B2-F1-F1 was capable of identifying the protein band about 17 kDa (Fig. 3C).
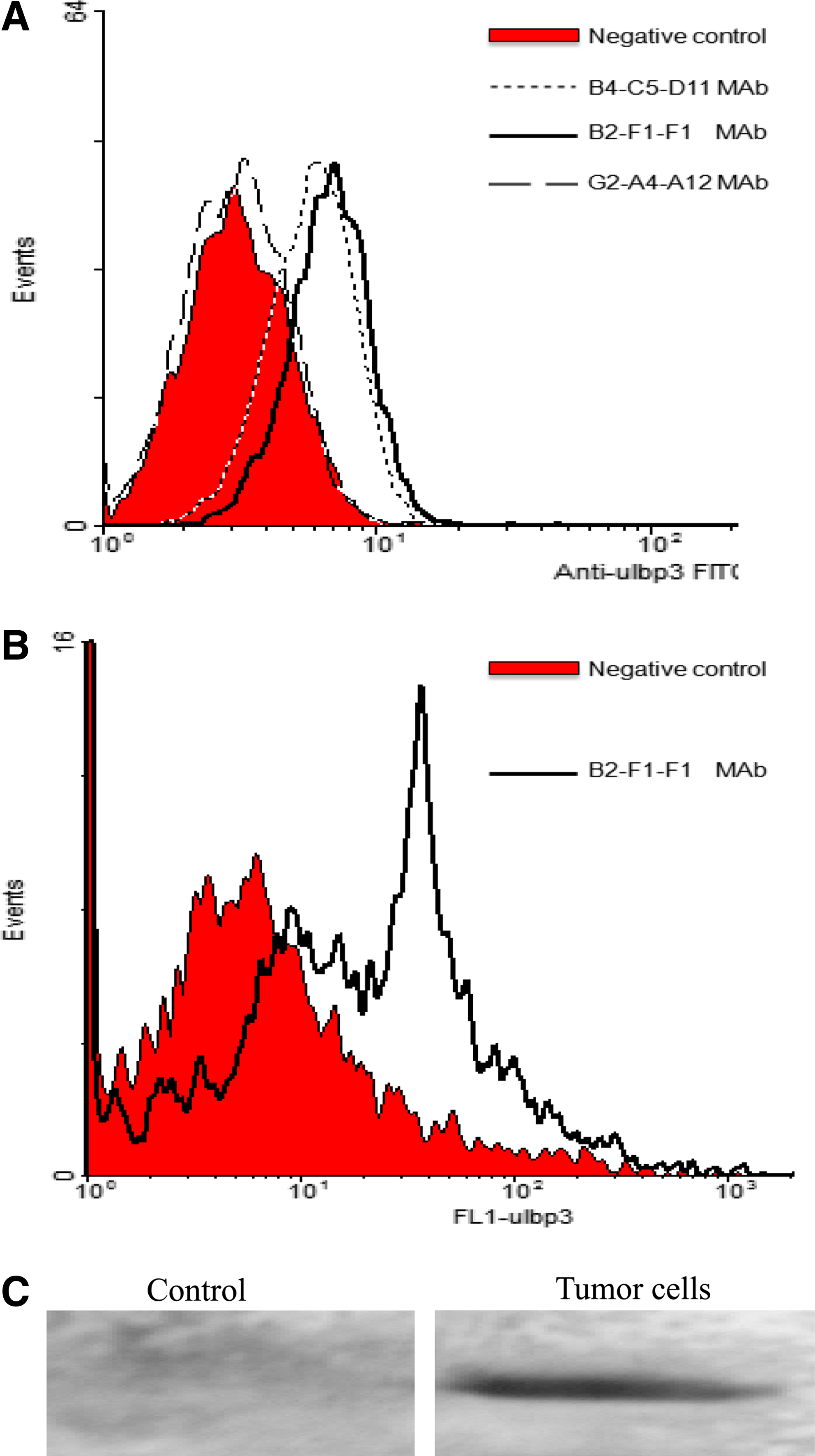
Binding affinity of anti-ULBP3 MAbs to nature ULBP3 on the cells. The cells from colorectal cancer tissue and SW620 lines were stained using anti-ULBP3 MAbs followed by FITC-conjugated goat anti-mouse IgG antibody as described in the Materials and Methods section. A total of 10,000 cells were analyzed in each experiment. (
Immunohistochemical detection of ULBP3 from colorectal cancer tissues
We wanted to know whether these anti-ULBP3 MAbs would also work successfully in immunohistochemistry applications. ULBP3 protein from colorectal cancer tissue sections was detected with the MAb (clone B2-F1-F1). The results showed that the tumor tissue specimens exhibited positive immunoreactivity with the MAb, and staining was on the plasma membrane (Fig. 4), indicating that the MAb may be applied in immunohistochemistry analysis.
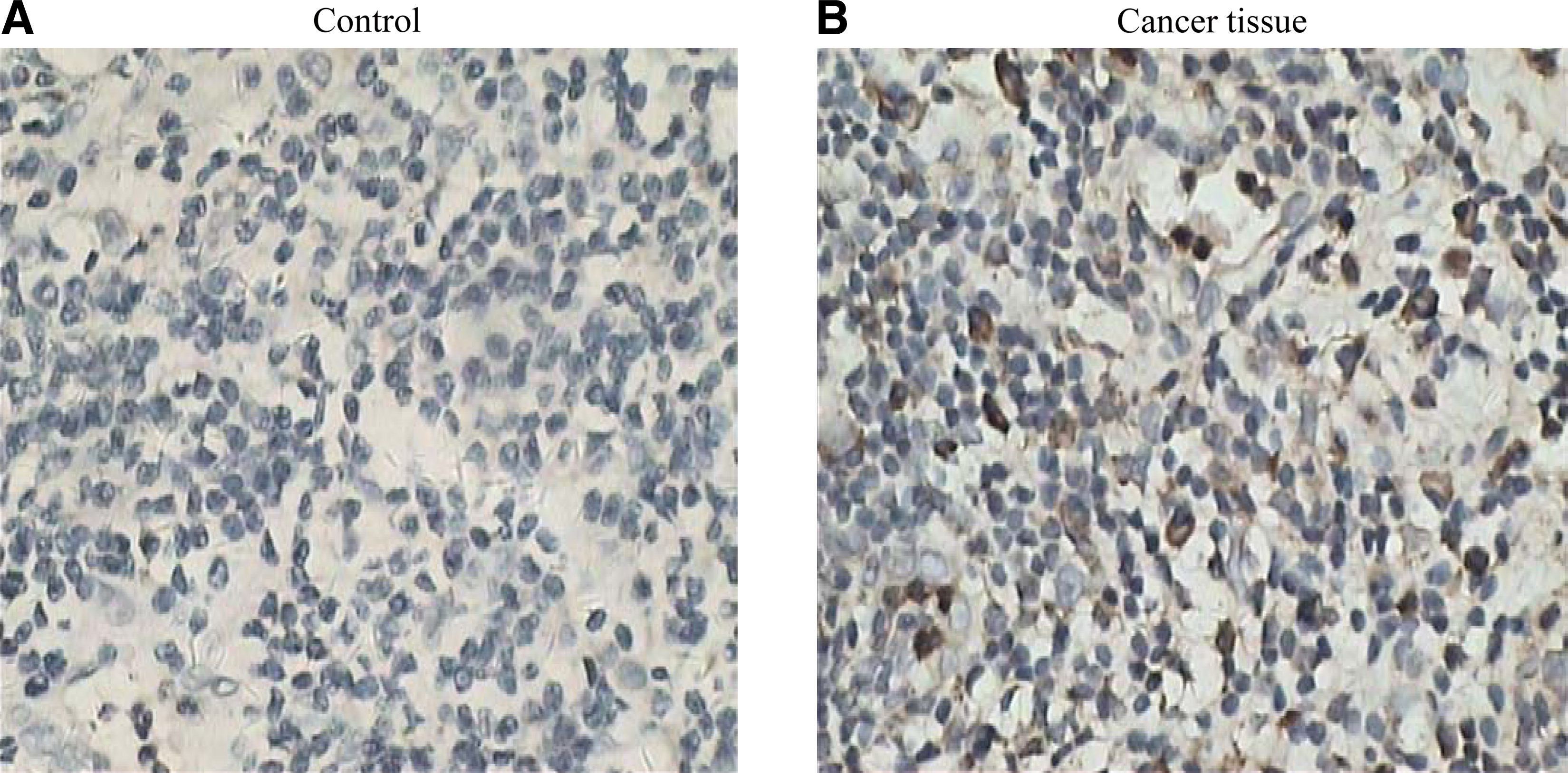
Detection of ULBP3 expression by immunohistochemical staining in the tumor tissue sections from colorectal cancer. Tumor tissues were immunostained using the prepared MAb (clone B2-F1-F1). Immunoreactivity was detected with HRP-labeled secondary antibody and diaminobenzidine (brown precipitate, 100×). Negative controls included sections immunostained with the primary antibody omitted or a non-relevant monoclonal antibody substituted for the anti-ULBP3 antibodies. (
Discussion
It is well-established that ULBP3 are known to bind to human NKG2D, which are expressed on NK cells, γδ T cells, and CD8+ T cells, resulting in the activation of cytotoxic activity and cytokines production by these effector cells. In this study, we used rec-ULBP3 as antigen and produced three MAbs against ULBP3 by hybridoma technology and demonstrated that MAbs from clones B2-F1-F1 and B4-C5-D11 could recognize nature ULBP3 on eukaryotic cell line SW620 and colorectal cancer cells and may work in immunohistochemistry applications. Thus, these prepared anti-ULBP3 MAbs provide a potential tool for further understanding the physiological and pathological role of ULBP3.
The specificity of the prepared MAbs is critical. In this study, we not only proved the expression of ULBP3 on the colorectal cancer cell line SW620 by PCR analysis, but also examined the specific affinity of these prepared MAbs to nature ULBP3. This will set the stage for clinical application of the MAb against ULBP3. Based on the intra/extracellular expression patterns of ULBPs in some tumor cells, it seems that ULBPs are synthesized and exported to the cell surface, where they are cut by phosphatidylinositol-specific phospholipase C to be released as soluble forms. Some soluble ULBPs are mainly responsible for down-regulating NKG2D expression of NK cells in gastric tumors.(20,27) Next, we wanted to establish a method for detecting the concentration of soluble serum ULBP3 in tumor patients in order to determine whether serum ULBP3 may serve as a type of tumor marker. In addition, we also explored the relationship between NKG2D and soluble ULBP3 in the immune escape of tumor patients by means of these prepared MAbs against ULBP3.
As a result, we generated a recombinant protein ULBP3 and successfully prepared MAbs against ULBP3. These MAbs can recognize human ULBP3 protein and are suitable for flow cytometric analysis and immunohistochemical staining, which provide a valuable tool for further investigation of ULBP3 function and clinical application.
Footnotes
Acknowledgments
We thank Yancai Wei for his help in preparing the rec-ULBP3. This work was supported by the Clinical Medicine Foundation of Jiangsu University (JLY2010157), the Natural Science Foundation of Jiangsu Province of China (BK2009208), and the Science and Technology Commission of Zhenjiang Municipality (SH2011020, SH2008034).
Author Disclosure Statement
The authors have no financial interests to declare.