
Abstract
A proteasome is a multi-subunit protein complex, which plays a central role in ubiquitin-dependent protein degradation in all eukaryotic cells. The 26S proteasome is composed of a catalytic 20S core complex and one or two 19S regulatory complexes. The 20S core complex forms a cylinder consisting of four stacked rings of seven α (PSMA1-7) or β (PSMB1-7) subunits. Target proteins are degraded in the cavity of the 20S complex due to proteolytic activities of three β subunits having catalytic sites located on the inner surface of the cylinder. The aim of this study was the generation of polyclonal antibodies against human proteasome subunits PSMA3, PSMA5, and PSMB5 and characterization of their experimental applications. To construct GST-fusion proteins, DNA sequences encoding PSMA3, PSMA5, and PSMB5 were cloned into prokaryotic expression vectors pGEX-5X-1 or pGEX-4T-3. Recombinant proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 were highly expressed in E. coli BL21 (DE3) cells, purified by glutathione-affinity chromatography and further used for rabbit immunization. The activity and specificity of the obtained antibody-containing sera were evaluated using Western blot analysis and immunoprecipitation. We have shown by Western blot analysis that our anti-PSMA3, anti-PSMA5, and anti-PSMB5 antibodies recognized both recombinant and endogenous proteins from different human cell lines. We have also shown that anti-PSMA3 and anti-PSMA5 sera were able to recognize and immunoprecipitate native forms of both endogenous and overexpressed FLAG-tagged proteins PSMA3 and PSMA5, respectively. Thus, the antibodies generated against PSMA3, PSMA5, and PSMB5 can be used in various experimental applications, including the evaluation of cellular levels of proteasome subunits in cell extracts and affinity purification of the endogenous and/or overexpressed proteasome subunits, which facilitates subsequent analysis of their post-translational modifications as well as protein-protein interactions in vivo.
Introduction
In recent work we have revealed that the 26S proteasome exhibits endoribonuclease activity and that PSMA5 is its catalytic RNase subunit. Moreover, experimental data indicated that an endoribonuclease activity of the 26S proteasome could be controlled by phosphorylation of the two other 20S CP subunits, PSMA1 and PSMA3.(6) It has also been shown by our group that the 20S proteasome is involved in the regulation of alternative splicing in vitro, and that PSMA3 interacts directly in vitro with proteins that are involved in various aspects of mRNA metabolism, including splicing.(7)
Here we describe the generation of polyclonal antibodies against human proteasome subunits PSMA3, PSMA5, and PSMB5 and characterize their experimental applications. The generated antibodies can serve as a powerful tool for further functional studies of the 20S CP subunits, particularly in understanding the molecular mechanisms underlying the regulation of mRNA metabolism by PSMA3 and PSMA5 proteins.
Materials and Methods
Materials
Escherichia coli DH10 cells were used for cloning and plasmid amplification, and E. coli BL21 (DE3) cells were used for the expression of GST-fused protein. Human cell lines HeLa, 293T, and H1299 were obtained from the cell culture bank of the Institute of Cytology, RAS (St. Petersburg, Russia). The following plasmids were used: pGEX-5X-1, pGEX-4T-3 (GE Healthcare, Uppsala, Sweden), and pFLAG-CMV-4 (Invitrogen, Carlsbad, CA). The following antibodies were used for Western blot analysis: mouse anti-FLAG (M2, (Sigma-Aldrich, St. Louis, MO); mouse anti-PSMA3 (Enzo Life Sciences, Farmingdale, NY); and mouse anti-PSMA5 (Enzo Life Sciences). HRP-conjugated goat anti-rabbit and rabbit anti-mouse antibodies were purchased from Sigma-Aldrich. DNA modifying and restriction enzymes were obtained from Fermentas (Glen Burnie, MD) or New England BioLabs (Ipswich, MA). The oligonucleotide synthesis was carried out by Sigma-Aldrich.
Cloning and generation of expression vectors
To generate vectors for the expression of the GST-fused proteasome subunits in E. coli BL21 (DE3) DNA sequences encoding PSMA3, PSMA5, and PSMB5 (GenBank accession nos. NM002788, NM002790 and NM_002797, respectively) were amplified from a cDNA library of human K562 cells by using the following primers: GST-PSMA3-F 5′-ATGC
To generate expression vectors for the FLAG-tagged proteasome subunits in human cells, DNA sequences encoding PSMA3, PSMA5, and PSMB5 were amplified from a cDNA library of human K562 cells by use of the following primers: FLAG-PSMA3-F 5′-GG
Competent E. coli DH10 cells were transformed with obtained recombinant plasmids pGEX-PSMA3, pGEX-PSMA5, pGEX-PSMB5, pFLAG-CMV-PSMA3, pFLAG-CMV-PSMA5, or pFLAG-CMV-PSMB5 and plated onto LB agar plates supplemented with ampicillin (100 μg/mL). Transformants were screened by PCR and were subsequently verified by the restriction analysis. The accuracy of cloning was confirmed by sequencing.
Expression of GST-fused proteins in E. coli
E. coli BL21 (DE3) cells were transformed with pGEX-PSMA3, pGEX-PSMA5, or pGEX-PSMB5 vectors encoding GST-PSMA3, GST-PSMA5, and GST-PSMB5 proteins, respectively. Transformed cells were plated onto LB agar plates supplemented with ampicillin (100 μg/mL). To express GST-fused proteins, transformants were grown overnight at 37°C in 3 mL of LB medium containing 100 μg/mL ampicillin. The overnight cultures were used to inoculate 250 mL of LB medium containing 100 μg/mL ampicillin. When the OD600 nm reached 0.5–0.6, protein expression was induced by adding isopropyl-β-
Affinity purification of GST-fused proteins
The solubility of the expressed proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 was determined before purification. The cell pellets were resuspended in 4 mL buffer LB containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mg/mL lysozyme, 5 mM DTT, 1 mM EDTA, and 0.5 mM PMSF. The obtained cell suspensions were sonicated on ice and centrifuged at 15,000 g for 30 min at 4°C. The supernatants and pellets were analyzed by SDS-PAGE with subsequent Coomassie Blue staining. Soluble GST-PSMA3 and GST-PSMB5 proteins were affinity-purified by batch method using glutathione sepharose (GS) beads (GE Healthcare, Uppsala, Sweden). The supernatants containing soluble fractions of GST-PSMA3 or GST-PSMB5 proteins were agitated with 200 μL of pre-equilibrated GS beads for 40 min at 4°C. The beads were then washed five times with 800 μL washing buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM DTT, 1 mM EDTA, 0.5 mM PMSF). GST-PSMA3 and GST-PSMB5 proteins were then eluted with buffer containing 50 mM Tris-HCl (pH 8.5), 150 mM NaCl, 50 mM glutathione, 5 mM DTT, and 0.5 mM PMSF.
The pellet containing insoluble GST-PSMA5 protein was resuspended in 4 mL LB buffer supplemented with 1.3% Sarkosyl and incubated for 10 min on ice. Then Triton X-100 was added to a final concentration of 2.6%. The following incubation with GS beads and later purification were performed under the same condition as was done for soluble proteins. The efficiency of purification and the purity of eluted proteins were analyzed by SDS-PAGE, followed by Coomassie blue staining. Final concentration of the eluted proteins was estimated by Bradford protein assay.(8) Samples were aliquoted and stored at −80°C until further immunization.
Generation of polyclonal antibodies against the recombinant proteins
The purified recombinant proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 were used for the generation of antibodies in rabbits.(9) Three rabbits were first immunized subcutaneously with 100 μg of each recombinant protein in complete Freund's adjuvant. Three weeks later two sequential booster injections were done with 100 μg of recombinant proteins in incomplete Freund's adjuvant at 4-week intervals. Blood was collected 8 days after the last immunization and serum containing specific antibodies was obtained. Control pre-immune serum was obtained from the same rabbits before immunization.
SDS-PAGE and Western blot analysis
All samples were prepared by boiling in a Laemmli buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 0.05% bromophenol blue, 10% glycerol, 100 mM DTT) for 7 min.(10) Then proteins were resolved by 12% SDS-PAGE followed by visualization using Coomassie blue staining. For Western blotting, proteins were transferred onto a PVDF membrane after separation by SDS-PAGE. The obtained membrane was blocked with PBS supplemented with 5% (w/v) non-fat dry milk and 0.1% (v/v) Tween-20 for 1 h at room temperature (RT) or overnight at 4°C. The following incubation of the membrane with appropriate primary and HRP-conjugated secondary antibodies was performed in PBS supplemented with 1% (w/v) non-fat dry milk and 0.1% (v/v) Tween-20 for 1 h at RT. The membranes were washed six times in PBS supplemented with 0.1% (v/v) Tween 20 after incubations with primary and secondary antibodies. ECL detection was-performed with SuperSignal system (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol.
Immunoprecipitation
Non-transfected and transiently transfected 293T cells with vectors pFLAG-CMV-PSMA3 or pFLAG-CMV-PSMA5 were washed twice with ice-cold PBS and lysed in the lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail [Roche, Mannheim, Germany]) by agitation for 30 min at 4°C. The resulting cell lysate was clarified by centrifugation at 20,000 g for 30 min at 4°C, and the supernatant was collected. The cell extract was pre-incubated with Protein A–Sepharose (GE Healthcare) for 1 h at 4°C. Upon preincubation, it was incubated with antibodies anti-PSMA3 or anti-PSMA5 (dilution 1:100) conjugated to Protein A–Sepharose for 4 h or overnight at 4°C under gentle rocking. The beads were then pelleted by centrifugation and washed five times with buffer containing 50 mM Tris-HCl (pH 8.0) and 150 mM NaCl. Immunoprecipitated proteins were analyzed by Western blot analysis using commercial mouse antibodies against PSMA3 or against PSMA5.
Cell culture and transfection
HeLa, 293T, or H1299 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS (Gibco, Carlsbad, CA), 2 mM
Transfection of 293T cells with vectors pFLAG-CMV-PSMA3 or pFLAG-CMV-PSMA5 was done using the calcium phosphate method.(11) Briefly, cells were seeded in a 60 mm culture plate and then grown to 50–70% confluence. One h before transfection, fresh medium (4 mL) was added to cells. Pure plasmid DNA (3–4 μg) was dissolved in 200 μL of 250 mM CaCl2 solution. Then the mixture was carefully added to 200 μL of 2x HBS buffer (50 mM Hepes, 10 mM KCl, 1.5 mM Na2HPO4, and 280 mM NaCl [pH 7.05]) under concomitant vortexing. After 20 min incubation at room temperature, the mixture (final volume of 400 μL) was added dropwise onto the culture plate. The culture plate was carefully shaken to equally disperse the calcium phosphate precipitates. Cells were incubated for 36–48 h at 37°C, and then the whole cell extract was prepared and used for immunoprecipitation or Western blot analysis.
Results and Discussion
The aim of this study was the generation of polyclonal antibodies against human proteasome subunits PSMA3, PSMA5, and PSMB5. Fusion of a protein of interest with glutathione S-transferase and expression of the fused protein in E. coli is a popular experimental approach that allows production and purification of proteins that can be used for further animal immunization.(12)
As a first step, PSMA3, PSMA5, and PSMB5 genes were amplified using specific primers from a human cDNA library. The corresponding PCR fragments of PSMA3 and PSMA5 were then cloned into the vector pGEX-5X-1, whereas PSMB5 fragment was cloned into the vector pGEX-4T-3. The obtained recombinant vectors with N-terminal GST, pGEX-PSMA3, pGEX-PSMA5, and pGEX-PSMB5 were identified by PCR screening and restriction analysis and then confirmed by sequencing.
To express the fusion proteins GST-PSMA3, GST-PSMA5, and GST-PSMB E. coli BL21 (DE3) cells were transformed with vectors pGEX-PSMA3, pGEX-PSMA5, and pGEX-PSMB5, respectively. Expression of the fusion proteins was induced by the addition of IPTG at 30°C for 3 h. To estimate the levels of expression for each protein whole cell lysates were obtained before and after induction with IPTG and were subsequently analyzed using SDS–PAGE followed by staining with Coomassie Blue. As evident from Figure 1, recombinant proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 were expressed as major products in cell lysates and migrated in the gel as single bands of expected molecular weights: 55 kDa, 53 kDa, and 55 kDa, respectively.
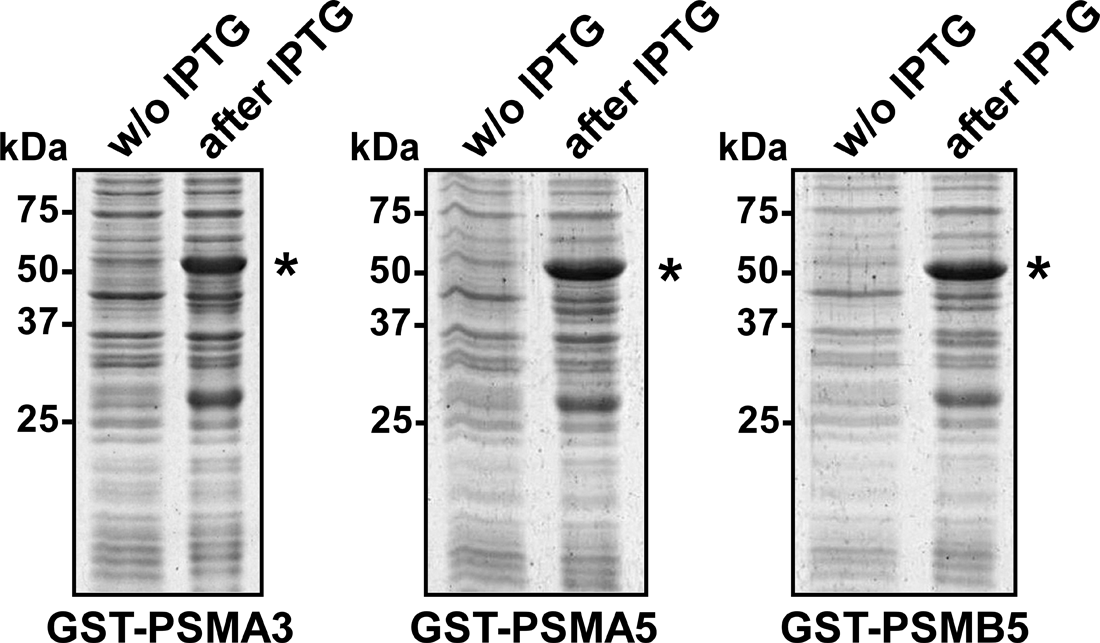
Expression of the fusion proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 in E. coli. Expression of GST-fused proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 in E. coli (BL21) cells transformed with the corresponding expression vectors after induction with IPTG. Whole lysates from control (w/o IPTG) cells or cells expressing GST-fused proteins (after IPTG) were analyzed by SDS–PAGE. Asterisks indicate the positions of the hybrid proteins GST-PSMA3, GST-PSMA5, or GST-PSMB5.
To purify overexpressed GST-fused proteasome subunits, we applied affinity chromatography based on the binding of GST-fused proteins with glutathione sepharose matrix.(13) To choose the appropriate strategy for the purification of expressed proteins GST-PSMA3, GST-PSMA5, or GST-PSMB5, we first evaluated their solubility. The supernatants and pellets obtained from bacterial whole cell lysates were analyzed using SDS–PAGE followed by Coomassie Blue staining. We detected the GST-PSMA3 protein only in the soluble fraction, whereas the GST-PSMA5 protein was found in the pellet fraction, suggesting that GST-PSMA3 was soluble and GST-PSMA5 was insoluble. The fused protein GST-PSMB5 had a mixed distribution being partly in the insoluble fraction and to a lesser extent in the soluble fraction (Fig. 2).
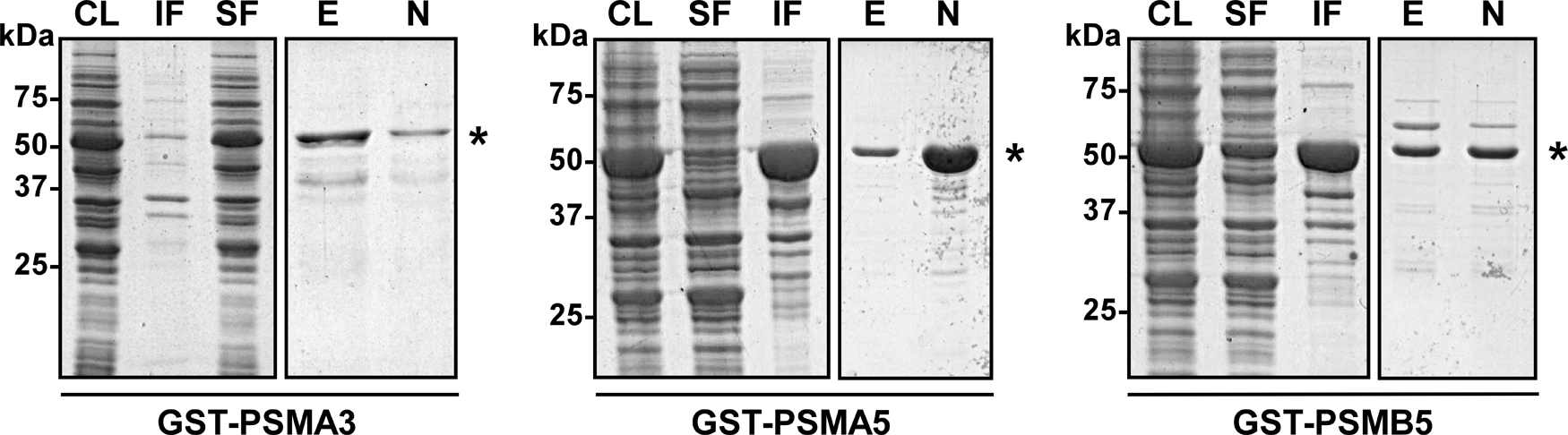
Examination of solubility and purification of the fusion proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5.Whole cell lysates (CL) from cells containing GST-fused proteins were sonicated and then centrifuged. The supernatants corresponding to the soluble fraction of proteins (SF) were collected. Proteins from the pellets corresponding to the insoluble fraction (IF) were solubilized by treatment with 1.3% Sarkosyl. GST-fused proteins were then purified by binding to glutathione sepharose (GS) beads followed by intensive washing. Sarkosyl-treated samples were supplemented with 2.6% Triton X-100 before incubation with GS beads. GST-fused proteins were then eluted by incubation of GS beads with an elution buffer containing glutathione. Fractions of eluted (E) and non-eluted (N) proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 were collected. All fractions were analyzed by SDS–PAGE. Asterisks indicate the positions of the hybrid proteins GST-PSMA3, GST-PSMA5, or GST-PSMB5.
Soluble proteins GST-PSMA3 and GST-PSMB5 were affinity-purified by batch method using GS beads from the corresponding supernatants. To purify the insoluble protein GST-PSMA5 from the pellet fraction, we used a modified experimental approach. Before binding to GS beads, insoluble proteins were solubilized by adding an ionic detergent Sarkosyl, followed by the addition of a non-ionic detergent Triton X-100. The latter forms micelles and thus sequesters away the ionic detergent from the protein, while still keeping it soluble. It has been shown that this treatment can maintain soluble fused proteins without affecting their ability to bind to affinity matrix.(14,15) After binding to GS beads and intensive washing, fused proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 were eluted from affinity matrix with glutathione. Purified proteins were analyzed using SDS–PAGE and subsequent Coomassie Blue staining. As shown in Figure 2, all three fused proteins GST-PSMA3, GST-PSMA5, and GST-PSMB5 were successfully purified. The yield of each purified recombinant protein was about 0.8 mg per 500 mL of bacterial culture, which is sufficient for several sequential rabbit immunizations.
To generate specific antibodies to proteasome subunits PSMA3, PSMA5, and PSMB5, three rabbits were immunized with the corresponding purified proteins. 100 μg of each fusion protein were used for three sequential immunizations (as described in the Materials and Methods section), and sera containing polyclonal antibodies were obtained. To examine the specificity of anti-PSMA3, anti-PSMA5, and anti-PSMB5 polyclonal antibodies, we first demonstrated that they were able to recognize their respective antigens by Western blot analysis. Ten ng of either purified protein GST-PSMA3, or GST-PSMA5, or GST-PSMB5 was subjected to SDS–PAGE and analyzed by Western blot analysis using the corresponding antibodies: anti-PSMA3 (dilution 1:1000), anti-PSMA5 (dilution 1:1000), or anti-PSMB5 (dilution 1:300). As shown in Figure 3, signals corresponding to the fusion proteins GST-PSMA3 (55 kDa), GST-PSMA5 (53 kDa), and GST-PSMB5 (55 kDa), and GST protein (26 kDa) were detected. No signal was detected when the same samples were incubated with control pre-immune serum (data not shown).
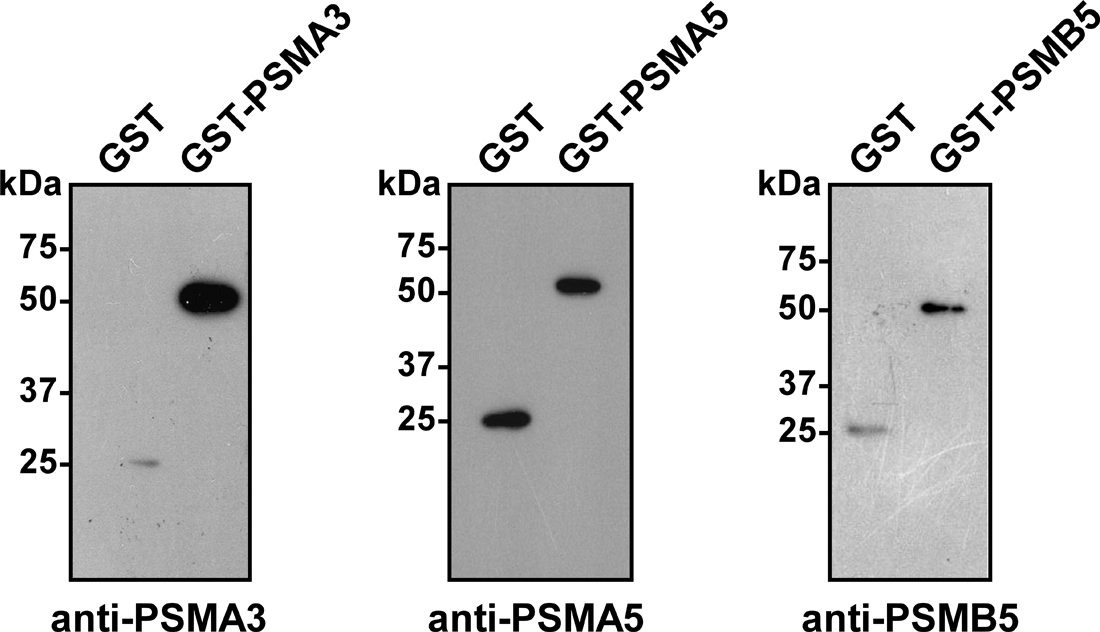
Generated antibodies to PSMA3, PSMA5, and PSMB5 recognize corresponding antigens. Purified fused proteins GST-PSMA3, GST-PSMA5, GST-PSMB5, and/or GST were separated by SDS–PAGE and analyzed by Western blotting using anti-PSMA3, anti-PSMA5, or anti-PSMB5 sera.
These antibodies were then tested for their ability to recognize the endogenous proteasome subunits PSMA3, PSMA5, and PSMB5 in whole-cell extracts of different human cells. Bands of expected sizes (28.4 kDa for PSMA3 and 26.4 kDa for PSMA5 proteins) were detected in HeLa and 293T cells by Western blot analysis using anti-PSMA3 (dilution 1:1000) and anti-PSMA5 (dilution 1:1000) sera, respectively (Fig. 4A, B, left panels). Signals corresponding to PSMB5 (21.8 kDa) were detected in 293T and H1299 cells using anti-PSMB5 antibodies (dilution 1:300) (Fig. 4C, left panel). Predicted molecular weight of the full-length PSMB5 subunit is 29 kDa, but it has been shown that N-terminal propeptide of the PSBM5 protein is autocatalytically cleaved during the assembly of 20S proteasome.(16) Thus, our antibodies detected cleaved form of PSMB5. The same samples were also incubated with the control pre-immune serum but no signal was observed (data not shown).
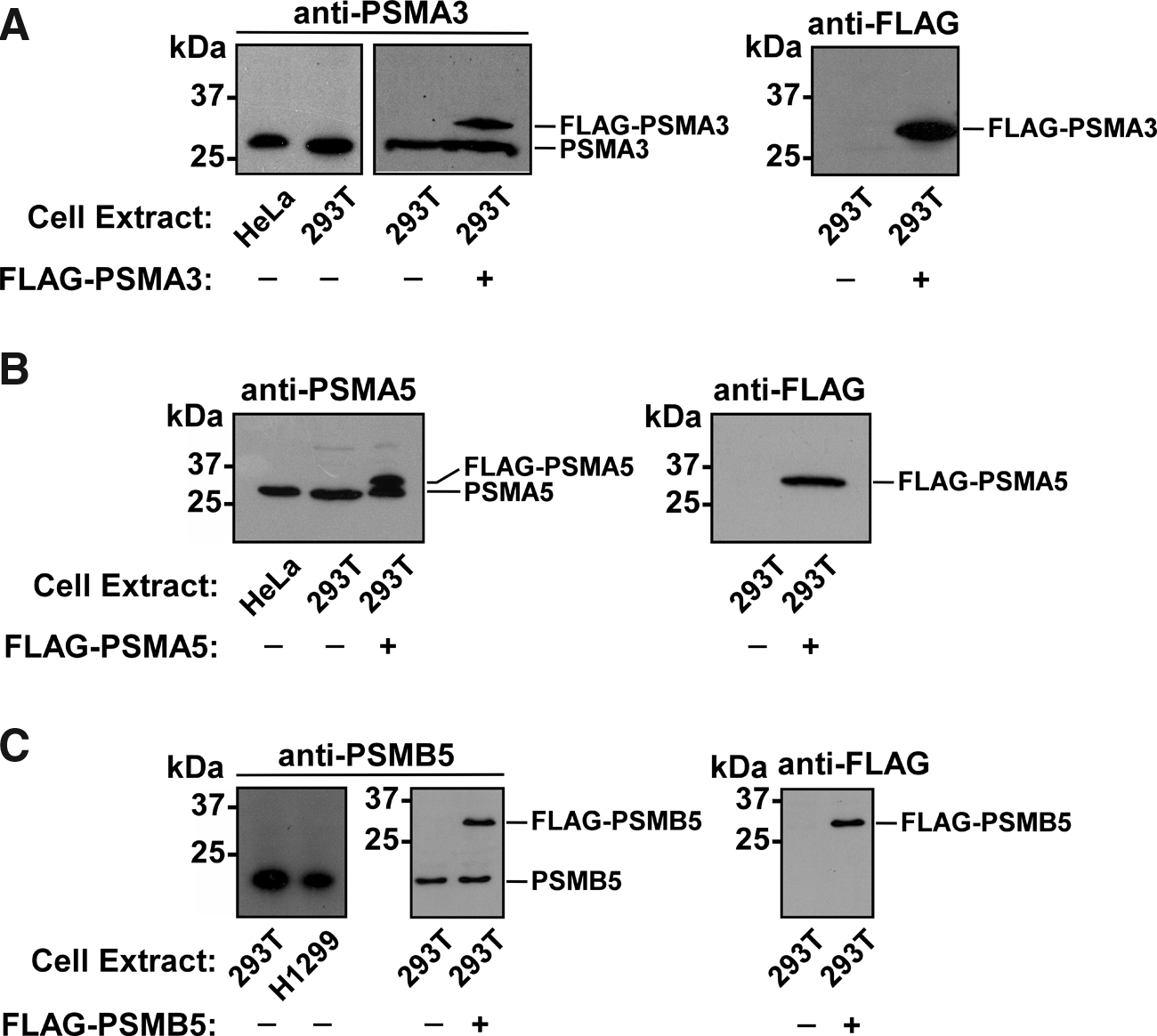
Antibodies to PSMA3, PSMA5, and PSMB5 recognize corresponding endogenous proteins from human cells. Whole extracts of HeLa, H1299, and 293T cells transiently transfected with vectors encoding FLAG-tagged human proteins PSMA3, PSMA5, or PSMB5 were separated by SDS–PAGE and analyzed by Western blotting using the following antibodies generated by us: anti-PSMA3 (
To test if our anti-PSMA3, anti-PSMA5, and anti-PSMB5 antibodies were able to recognize the corresponding proteins overexpressed in human cell lines, we cloned DNA sequences encoding open reading frames of the PSMA3, PSMA5, and PSMB5 genes into the mammalian expression vector pFLAG-CMV-4, which contains an amino-terminally located FLAG epitope. 293T cells were transiently transfected with pFLAG-CMV-PSMA3, pFLAG-CMV-PSMA5, and pFLAG-CMV-PSMB5 plasmids. Whole cell extracts from control and transfected 293T cells were prepared and the quality of our antibodies was then examined by Western blot analysis. Our PSMA3-, PSMA5-, and PSMB5-specific antibodies detected both endogenous and ectopically expressed proteins in these lysates (Fig. 4A, middle panel; B, left panel; C, middle panel). Commercial mouse monoclonal antibodies against FLAG peptide were used as a positive control to detect overexpressed FLAG-PSMA3, FLAG-PSMA5, and FLAG-PSMB5 proteins in the same lysates (Fig.4–C, right panels).
Finally, we tested the ability of our antibodies to recognize native forms of the corresponding human proteasome subunits by immunoprecipitation. For this purpose 293T cells were transiently transfected with either pFLAG-CMV-PSMA3 or pFLAG-CMV-PSMA5 vectors. Transfected and non-transfected cells (used as control) were then lysed under non-denaturing conditions. Whole-cell extracts were prepared and subsequently incubated with antibodies against PSMA3 or PSMA5 (dilution 1:100) conjugated to Protein A–Sepharose beads. The beads were then intensively washed and the immunoprecipitated proteins bound to beads were analyzed by Western blot analysis using commercial mouse monoclonal antibodies against PSMA3 (Fig. 5A) or against PSMA5 (Fig. 5B). Both endogenous and overexpressed FLAG-tagged proteins PSMA3 and PSMA5 were successfully immunoprecipitated by our custom-made anti-PSMA3 and anti-PSMA5 antibodies, respectively (Fig. 5A, B). No immunoprecipitated proteins were detected when 293T cells extract was incubated with control pre-immune serum coupled to Protein A–Sepharose beads or with Protein A–Sepharose beads alone (Fig. 5A, B). As a control, whole cell extracts (WCE) of non-transfected and transfected 293T cells were used. Comparable levels of the endogenous PSMA3 and overexpressed FLAG-PSMA3 proteins were detected in whole cell extract of transiently transfected 293T cells using Western blot analysis with anti-PSMA3 antibodies (Fig. 5A). However, FLAG-PSMA3 was detected more dominantly than endogenous PSMA3 after immunoprecipitation from the same cell extract (Fig. 5A), suggesting that considerable portion of endogenous PSMA3 is incorporated into the 20S proteasome, thus masking the relevant epitope recognized by our antibodies; however FLAG-PSMA3 protein was mostly found in its unincorporated state and thus is available for recognition by anti-PSMA3 antibodies. Furthermore, our PSMB5-specific serum was not able to immunoprecipitate the endogenous PSMB5 protein from 293T cells extract (data not shown), probably because most of this protein is incorporated into the 20S proteasome.
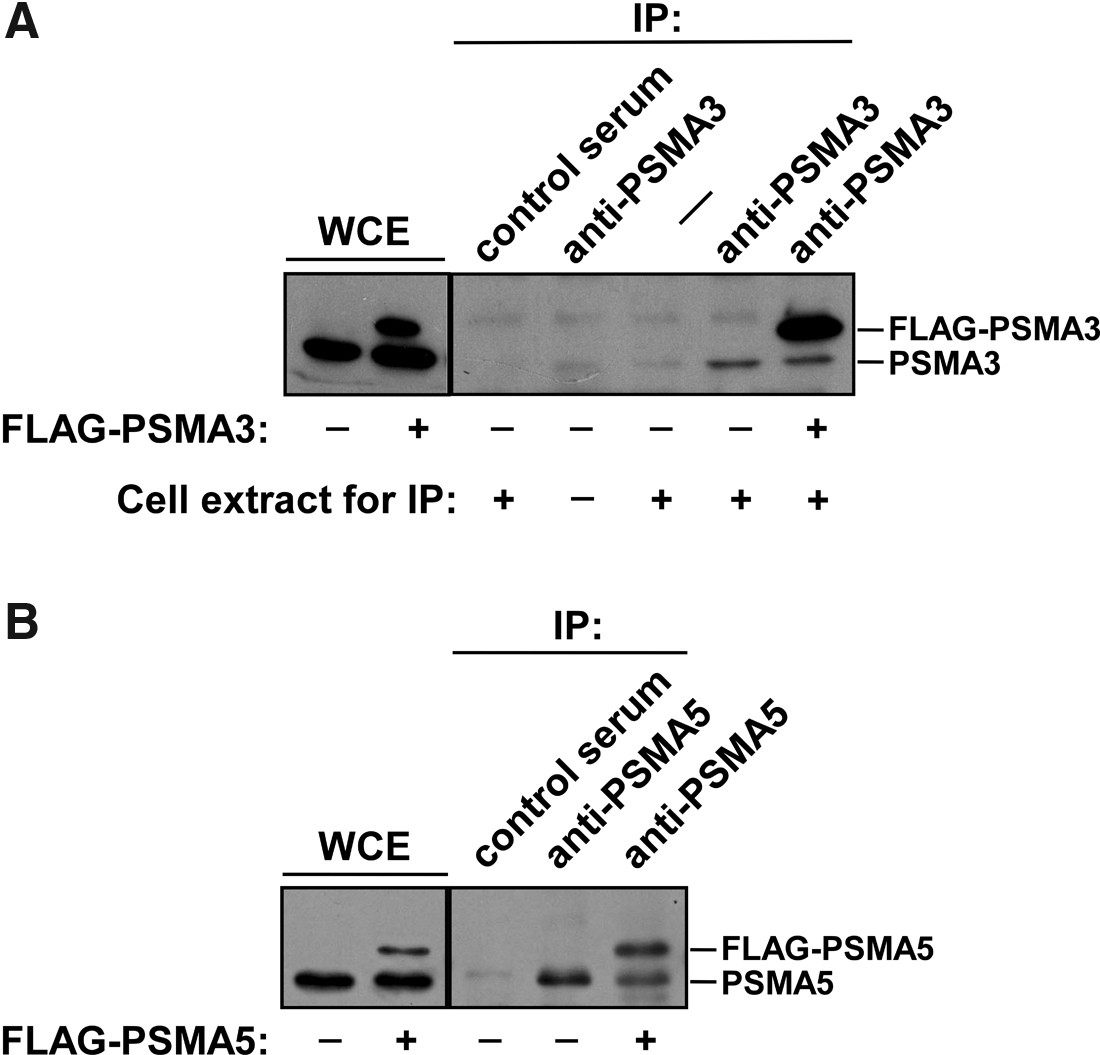
Antibodies to PSMA3 and PSMA5 immunoprecipitate endogenous and overexpressed human proteins PSMA3 and PSMA5. 293T cells were transiently transfected with vectors encoding FLAG-tagged human proteins PSMA3 or PSMA5. Control or transfected 293T cells were lysed under non-denaturing conditions and the endogenous or FLAG-tagged proteins PSMA3 or PSMA5 were immunoprecipitated using our anti-PSMA3 or anti-PSMA5 sera, coupled to Protein A–Sepharose beads. Western blot analysis demonstrated the presence of PSMA3 (
Thus, we successfully generated specific antibodies against PSMA3 and PSMA5 (anti-PSMA3 and anti-PSMA5), which can be used for affinity purification of the endogenous and/or overexpressed proteasome subunits to determine their post-translational modifications, as well as protein-protein interactions in vivo.
Footnotes
Acknowledgments
This work was supported by grants to N.B. from AICR, MCB RAS, RFBR (10-04-01234, 12-04-01152-9), The Program of the Ministry of Education & Science FASI (nos. 16.740.11.0366 and 14.740.11.0920). The work was performed using scientific equipment of the center of Shared Usage, “The Analytical Center of Nano- and Biotechnologies of SPbSPU.
Author Disclosure Statement
The authors have no financial interests to disclose.