
Abstract
Development of a specific immunoassay to detect Helicobacter pylori infection in stool samples requires monoclonal antibody against the specific antigen. The aims of this study were to establish monoclonal antibodies against the 26 kDa protein of H. pylori and develop an immunodot blot for their application to recognize H. pylori infection using stool samples. Mice were immunized intraperitoneally with homogenized gel containing the 26 kDa band of cell surface proteins of H. pylori in sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The monoclonal antibodies were produced using the hybridoma technique. Reactivity of monoclonal antibodies was tested with the purified 26 kDa antigen and cell surface proteins from cultured H. pylori by ELISA. Furthermore reactivity of monoclonal antibodies was tested on negative and positive stool samples for H. pylori and suspensions of several major bacteria in stool by immunodot blot assay. Five stable hybridoma monoclones were obtained. The concordant reactivity of the monoclonal antibodies with H. pylori present in the stool samples, which had been tested previously using an ACON ELISA kit for H. pylori stool antigen testing, and unreactivity with several different major fecal bacteria in immunodot blotting indicates high specificity of the immunodot blot based on the reaction of produced monoclonal antibodies with the H. pylori antigen in stools. The findings indicate that the novel immunodot blot developed based on new monoclonal antibodies for stool antigens would be useful as a noninvasive method of diagnosing H. pylori infection.
Introduction
Among several H. pylori antigens that have been isolated from the stools of infected people, the 26 kDa protein has a higher concentration and has been seen in nearly all positive samples.(23) It has also been introduced as a H. pylori specific antigen that is antigenically conserved and has been suggested to be useful as a diagnostic antigen in enzyme immunoassay tests to detect H. pylori infection.(24–26) Lundström and Bölin reported that the 26 kDa protein of H. pylori showed alkyl hydroperoxidase activity.(27) In addition to the evidence mentioned above, due to the possibility of AhpC of H. pylori as a prognostic or diagnostic protein marker to monitor varied clinical manifestations of gastrointestinal patients infected with H. pylori,(4) the 26 kDa protein of H. pylori was chosen as an antigen. In this report we describe the preparation and characterization of monoclonal antibodies (MAbs) against the 26 kDa protein (AhpC) and the development of an immunodot blot assay that allows specific identification of H. pylori infection.
Materials and Methods
Bacterial isolation and cultivation
The H. pylori strains used in this study were isolated from stomach biopsies in dyspeptic patients. The samples were placed directly on modified selective media containing Colombia Agar Base (Merck, Darmstadt, Germany) supplemented with 10% lysed horse blood, 0.25% yeast extract, 7% fetal calf serum (Gibco, Grand Island, NY), and M2 medium—amphotericin B (5 mg/L), trimethoprim (5 mg/L), and vancomycin (10 mg/L). Samples were scraped completely on the surface of the plates using a sterile loop. The cultures were kept in a microaerophilic atmosphere (7% O2, 7.1% Co2, 7.1% H2, and 79.8% N2) at 37°C. The plates were inspected first on day 1 and then on a daily basis for a total of 10 days. The isolates were confirmed as H. pylori by colony morphology, microscopic observation of spiral morphology, and positive oxidase, catalase, and rapid urease tests.(28) Bacterial cells from culture plates were harvested in phosphate-buffered saline (PBS, 10 mM, pH 7.2), washed twice in the same buffer and the pellet was stored at −20°C until use.
Extraction of cell surface proteins (CSPs)
An acid glycine extraction of CSPs of the Helicobacter species was prepared according to the method of Lelwata and colleagues.(29) Frozen cell pellets were thawed and cells were resuspended in 0.2 M glycine hydrochloride (pH 2.2; 4 g of cells/100 mL) supplemented with protease inhibitors (1.0 mM PMSF, 4 mM EDTA) and stirred magnetically for 15 min at 20°C. The suspension was centrifuged at 12000g for 15 min at 8°C, and the supernatant was neutralized with NaOH. Ammonium sulfate was added to 70% saturation to the supernatant. After 2 h incubation at 4°C, the extract was spun at 18000g for 30 min, and the pellet was resuspended in 3 mL PBS (10 mM, pH 7.2) and dialyzed for 18 h at 4°C against three changes of the same buffer. Protein was quantified by the Bradford method(30) and, after dividing into aliquots, kept frozen at −20°C until use.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
One-dimensional preparative and analytical sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) were performed using 12.5% separating gels and 5.0% stacking gels.(31) The protein extract (20 μL) was separated by analytical SDS-PAGE under reducing and non-reducing conditions and stained with Coomassie Brilliant Blue (CBB)-R250. The protein extract (0.75 mL) was also separated by preparative SDS-PAGE (180×160×1.5 mm) under reducing conditions and stained with zinc sulfate by the method of Bhattacharyya and colleagues.(32) Briefly, the gel was incubated for 15 min in 0.2 M imidazole and 0.1% SDS. It was then rinsed with distilled water and soaked in 0.2 M zinc sulphate for 1–2 min and finally rinsed with distilled water, which enabled the proteins to be visualized as transparent bands against a milky background.
Purification of the 26 kDa protein by electroelution
The designated band of the 26 kDa was separated from the preparative SDS-PAGE gels and cut into pieces using a scalpel blade. The gel pieces containing the appropriate protein were used for immunization and extraction of the protein using electroelution. Electroelution was performed as follows.
1. A 7-mm diameter hole was made through the head of a 5-mL syringe barrel.
2. The head was closed with paper tape and, while holding the barrel in a vertical position, 1 mL 1% molten agarose was poured into the barrel.
3. After the agarose was solidified, the pieces of SDS-PAGE gel were placed into the barrel.
4. An aliquot of 2 mL 1% molten agarose was poured into the barrel so that the pieces of the SDS-PAGE gel were embedded in agarose.
5. After the agarose solidified, a small dialysis bag with a 12-kDa cutoff containing 0.5 mL electrophoresis buffer was fastened around the head of the barrel and a similar dialysis bag was fastened around the bottom of the barrel.
6. The barrel was fixed horizontally in the chamber of a horizontal electrophoresis apparatus containing buffer consisting 25 mM Tris, 190 mM glycine, and 0.1% SDS (pH 8.3) such that the head of the barrel faced the positive pole.
7. Electroelution was performed overnight at 70 V and 4°C while the buffer was stirred.
8. At the end of electrophoresis, the polarity of the electrodes was changed for 30 s in order to avoid the adsorption of proteins to the dialysis bags.
9. The protein-containing buffer in the dialysis bags was collected.
10. For desalting and SDS removal, the solution collected was dialyzed against three changes of distilled water (pH 4.5) and then two changes of PBS (10 mM, pH 7.2). Protein was quantified by the Bradford method, divided into aliquots, and kept frozen at −20°C until use.
Preparation of monoclonal antibodies
Female BALB/c mice (6–8 weeks old) were immunized intraperitoneally (i.p.) with the homogenized gel containing approximately 10 μg of the 26 kDa antigen mixed with Freund's complete adjuvant for the first inoculation. Booster injections were administered i.p. with the homogenized gel containing ∼5 μg of the 26 kDa antigen mixed with Freund's incomplete adjuvant four times at intervals of 2 weeks. A final injection of 5 μg of the purified antigen without adjuvant was administered intravenously (i.v.). After 4 days, splenocytes and SP2/O myeloma cells were fused with 50% polyethylene glycol (PEG 1500). Hypoxanthine-aminopterine-thymidine (HAT) medium was used to select hybridoma cells. After 10 days, culture supernatants of hybridoma cells were screened for antibody production using indirect ELISA.(33,34) Microtiter plates were coated with 100 μL of the antigen 2 μg/mL of purified antigen in PBS) and incubated for 3 h at 37°C. Nonspecific binding sites were blocked with 100 μL of PBS containing 3% skim milk (blocking buffer) for 1 h at 37°C and then 100 μL of each hybridoma supernatant was added to each well. The plates were incubated for 1 h at 37°C, washed thrice with distilled water, and further incubated with 100 μL of an appropriate dilution of peroxidase-conjugated anti-mouse immunoglobulin G (HRP-anti-mouse IgG) for 1 h at 37°C. After five additional washings, 100 μL of substrate solution (0.64 mM tetramethylbenzidine dihydrochloride and 2.6 mM H2O2 in 0.2 M citrate buffer, pH 6.0) was added. The reaction was stopped after 15 min by the addition of 50 μL of 2 M HCl. All assay results were measured with a microplate reader at 450 nm. Positive hybrids were cloned twice by limiting dilution. The monoclones were cultivated for further analysis of secreted antibodies.
Isotyping of MAbs
The MAbs were isotyped using an IsoStrip™ Mouse Monoclonal Antibody Isotyping Kit (Roche, Mannheim, Germany).
Reactivities of MAbs with various bacteria by immunodot blotting
Cross-reactivity of MAbs with several kinds of major bacteria in stools were investigated. Bacterial strains to be tested (H. pylori, Escherichia coli, Shigella flexneri, Salmonella enteritidis, Vibrio cholerae, Yersinia enterocolitica, and Pseudomonas aeruginosa) according to the method of Bölin and colleagues(35) were suspended in phosphate-buffered saline (PBS 10 mM, pH 7.2) to an optical density of 1.5 at 600 nm. Samples of 2 μL of each suspension were applied to a nitrocellulose filter and air-dried for 15 min. The filter was blocked with 1% bovine serum albumin (BSA) in PBS for 30 min and then incubated with 1:10 dilution of MAbs in PBS containing 0.1% BSA and 0.05% Tween-20 (dilution buffer) for 1.5 h. The membrane was washed in washing buffer and incubated with the appropriate dilution of HRP-anti-mouse IgG in dilution buffer at room temperature for 1 h, and the peroxidase activity was developed in a substrate solution (1.4 mM 3, 3′-diaminobenzidine, 7.3 mM imidazole, and 0.889 mM H2O2 in 0.05 M Tris-HCl-buffered saline [pH 7.6]). Finally, the membrane was washed with distilled water.
Collection of stool samples
A total of 292 patients who had been referred to the clinical laboratory for H. pylori Stool Antigen test and had been already tested using the ACON ELISA kit were enrolled in this study. The stool samples were stored at −20°C until use. Patients enrolled in the study had not received antibiotics or acid suppressive drugs (proton pump inhibitors, H2 receptor antagonists, antacids, bismuth preparations) within 4 weeks prior to examination. The study was approved by the local ethics committee, and informed consent was obtained from all patients.
Preparation of fecal extract
The samples were thawed and thoroughly mixed. Then 0.3 g from each stool sample was suspended in 1 mL PBS (10 mM, pH 7.2) and the supernatant was obtained by centrifugation at 8000g for 10 min at room temperature.
Immunoblotting of fecal extracts
To estimate H. pylori AhpC molecular weight in the stool samples, 60 stool samples, which had previously been tested by immunodot blotting, were tested by immunoblotting. Analytical continuous SDS-PAGE of the samples was performed in 10% acrylamide gel. The resolved proteins were transferred to a nitrocellulose membrane. The membrane was blocked and incubated with a 1:10 dilution of MAbs in dilution buffer at room temperature for 1.5 h. After washing, the filter was developed with the appropriate enzyme conjugate and substrate as described for immunodot blotting.
Effect of low ionic strength on fecal extract
To consider the effect of low ionic strength on aggregated AhpC in stool samples, fecal extracts were dialyzed against three changes of distilled water and immunoblotting was performed as previously described.
Immunodot blotting of stool samples by MAbs
To evaluate the clinical application of the MAbs and development of a dot blot assay, a total of 292 stool samples from the patients were collected and fecal extracts prepared as previously described. An aliquot of 2 μL the fecal extract was dotted onto nitrocellulose membranes, which was incubated for 30 min at 37°C. The membranes were washed in PBS (10 mM, pH 7.2, containing 0.3% H2O2) with gentle agitation for 1 h, then blocked, treated with MAbs, washed, and developed by the same procedure as described for immunoblotting.
Determination of sensitivity in immunodot blotting
The protein of the purified H. pylori AhpC from stool was quantified, serially diluted in PBS (10 mM, pH 7.2), and tested using the MAbs in immunodot blotting.
Statistical analysis
Kappa statistics were used.
Results
SDS-PAGE analysis of CSPs extract
SDS-PAGE analysis of CSPs extract was performed under reducing and non-reducing conditions. In the presence of β-mercaptoethanol, as a reducing agent, in the sample buffer, the 26 kDa band was completely sharp and detectable, while in the absence of β-mercaptoethanol, the 26 kDa band was very weak and a strong band with molecular weight of 43 kDa was detected. These data suggest the presence of interchain disulfide bond in this protein (Fig. 1).
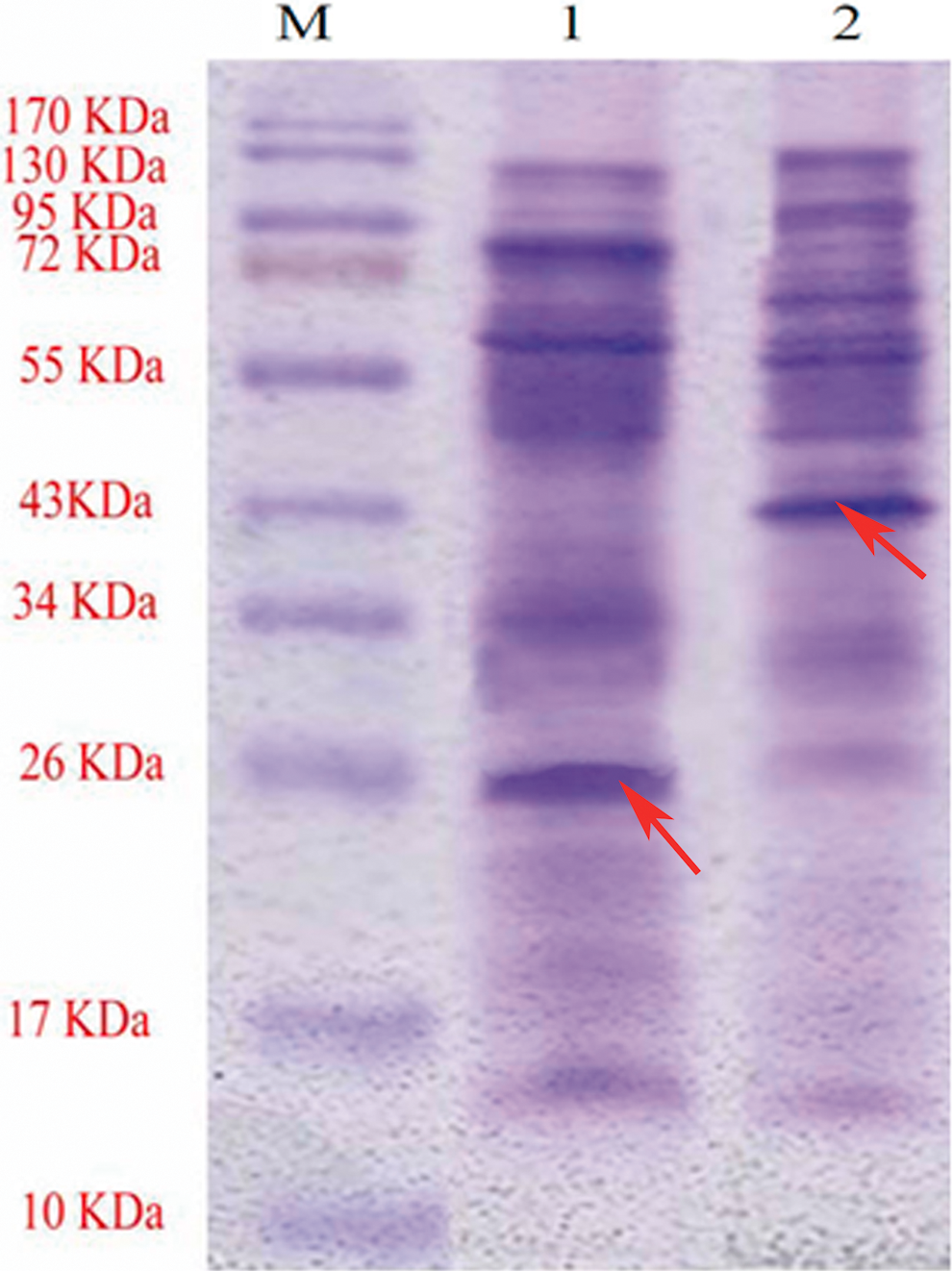
SDS-PAGE analysis of CSPs extract from H. pylori. Lane M, molecular weight markers. Lanes 1 and 2 represent the CSP extract, which was separated under reducing and non-reducing conditions respectively and was stained with Coomassie Brilliant Blue-R250. In the presence of β-mercaptoethanol in the sample buffer, the 26 kDa band was completely sharp and detectable while in the absence of β-mercaptoethanol, the 26 kDa band was very weak and a band with molecular weight of 43 kDa was detected. These data suggest the presence of interchain disulfide bond in this protein. The arrows indicate the position of bands.
Screening of MAbs
Among 42 hybridoma clones that reacted with the purified 26 kDa antigen and cell surface proteins of H. pylori by indirect ELISA, five stable hybridoma monoclones were obtained. These MAbs were designated A-hel26-A, A-hel26-B, A-hel26-C, A-hel26-D, and A-hel26-E.(36)
Isotyping of MAbs
MAb isotyping indicated that the clones all belonged to the IgG2 class and subclass. A-hel26-A, C, D, and E clones had kappa light chains and A-hel26-B clone had a lambda light chain.
Cross-reactivity of MAbs with several bacteria
Reactivities of the MAbs with various bacterial strains in immunodot blotting indicated a strong positive reaction of all MAbs with H. pylori and a very weak reaction with P. aeruginosa but not with E. coli, Shig. flexneri, Sal. enteritidis, V. cholera, and Y. enterocolitica (Fig. 2).

Reaction profiles of the MAb A-hel26-B by dot immunoblot analysis with various bacteria. Lanes 1, E. coli; 2, H. pylori; 3, Shig. flexneri; 4, Sal. enteritidis; 5, P. aeruginosa; 6, V. cholera; and 7, Y. enterocolitica. Positivity is confirmed by visual inspection of the brown color development in dots.
Immunoblotting
Studies on immunoblotting of stool extracts with MAbs (Fig. 3) and a polyclonal antibody to recombinant AhpC from H. pylori positive stools (Fig. 4)(37) indicated that in all cases the antigen gave a smeared and unclear band, with an approximate molecular weight of 200 kDa. Also, using freshly voided stools and dialysis of fecal extracts against distilled water to decrease the ionic strength did not change the position of the H. pylori antigen band on SDS-PAGE (data not shown).

Reaction profiles of MAb A-hel26-B immunoblot analysis with stool samples, which were identified as 35, 41, 42, 44, 47, and 48. Stool identification numbers are indicated above the lanes. Positivity is confirmed by visual inspection of the brown color development.
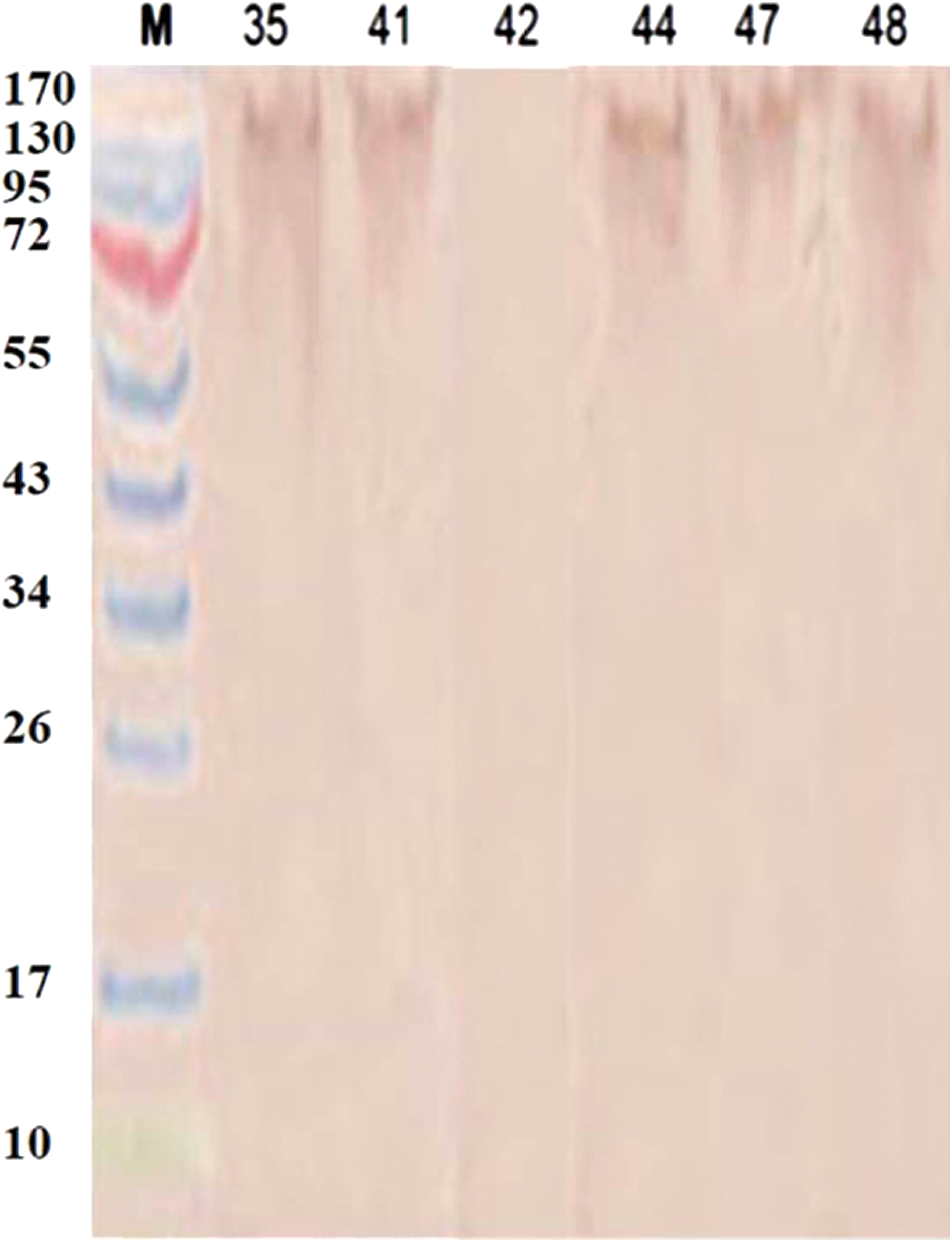
Reaction profiles of the polyclonal antibody to recombinant H. pylori AhpC in immunoblotting with stool samples that were identified as 35, 41, 42, 44, 47, and 48. Stool identification numbers are indicated above the lanes. Positivity is confirmed by visual inspection of the brown color development.
Comparison of immunodot blotting developed with ELISA in detecting H. pylori in stool
To estimate the clinical application of the MAbs, reactivity was tested by immunodot blot assay of 292 stool samples, which had been tested previously with the ELISA kit for H. pylori Stool Antigen testing (ACON Laboratories, San Diego, CA). Results of 69 samples are shown in Figure 5. Immunodot blot results of all MAbs showed high agreement with the ELISA kit results. Immunodot blot for MAb A-hel26-B and MAb A-hel26-A with the ELISA kit results were most concordant with each other (kappa coefficients 0.92 and 0.904, respectively). The highest discordance was observed between MAb A-hel26-D and the ELISA kit results (kappa coefficient 0.81). Results are summarized in Table 1.

Reaction profiles of the MAbs (A-hel26-A, B, C, D, and E) and PBS (as a negative control, N) by dot immunoblot analysis indicating 68 stool samples. Stool identification numbers are indicated above the dots. Positivity is confirmed by visual inspection of the brown color development in dots. The dots belonging to positive samples are brown while the dots belonging to negative samples are colorless or the same color of background in negative control.
n=292.
Sensitivity of immunodot blotting by MAbs
The lowest concentrations of H. pylori AhpC that could be detected by MAbs A-hel26-A, A-hel26-B, A-hel26-C, A-hel26-D, and A-hel26-E were 8 μg/mL, 8 μg/mL, 12 μg/mL, 20 μg/mL, and 16 μg/mL, respectively. Altogether, the detection limit of H. pylori AhpC for all MAbs was 8–20 μg/mL. Data for A-hel26-B are shown in Figure 6.
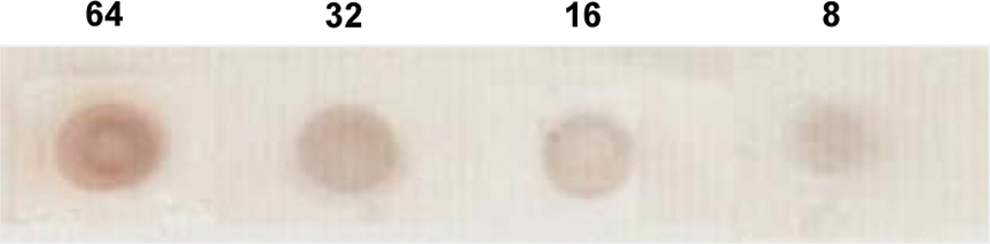
Sensitivity of immunodot blot assay for purified fecal H. pylori AhpC with the MAb A-hel26-B. Numbers above the dots indicate concentrations (μg/mL) of AhpC. Positivity is confirmed by visual inspection of the brown color development in dots.
Discussion
Several studies have reported the production and application of a MAb specific for H. pylori antigens,(35,38–42) but there are only a few reports of a MAb specific for a distinct fecal H.pylori antigen that can be used to detect antigens of H. pylori in stools. Two studies have reported the isolation of some H. pylori antigens from the stools of infected people. Suzuki and colleagues isolated a homotetramer antigen with a molecular weight of 260 kDa and a subunit molecular weight of 59 kDa. This antigen was identified as catalase.(16) Sheikhian and colleagues reported two major antigens of molecular weight 24–26 and 11.5–14.5 kDa that had been isolated from the stools of almost all infected patients in Iran.(23) The 26 kDa antigen was described as a species-specific protein of H. pylori by O'Toole and colleagues.(25) Huang and colleagues(4) suggested the possibility of AhpC of H. pylori as a prognostic or diagnostic protein marker to monitor varied clinical manifestations of gastrointestinal patients infected with H. pylori.
In the present study MAbs were produced against the 26 kDa protein of H. pylori. Antigen-containing gel from the SDS-PAGE (26 kDa band) and electroeluted purified 26 kDa protein were used as immunogen for i.p. and i.v. injections, respectively; purified protein was used for screening by ELISA as well. To stain the electrophoresis bands, zinc sulfate staining was used, which facilitates electrophoretic elution of H. pylori AhpC from SDS-PAGE gels, with no need to remove the dye. Purity of the protein was confirmed by performance of SDS-PAGE on the eluted protein. SDS is widely used for protein solubilization and for separation of proteins by SDS-PAGE; however SDS interferes with the indirect ELISA, protein quantification by Bradford method and also i.v. injection. In this study, SDS removal was performed efficiently through dialysis. Our studies with SDS-PAGE followed by immunoblotting of H. pylori AhpC from positive stool samples with MAbs indicated that the antigen band had an approximate molecular weight of 200 kDa with a smeared band. None of the positive stools for H. pylori AhpC showed a sharp 26 kDa band. Furthermore, the results of studies with a polyclonal antibody to recombinant AhpC from H. pylori were similar. In spite of the use of freshly voided stools and dialysis of fecal extracts against distilled water to decrease ionic strength, the position of the band of H. pylori AhpC on SDS-PAGE was not changed. These results suggest that the protein structure of AhpC probably shifts from low molecular weight oligomers with peroxide reductase activity to high molecular weight complexes with molecular chaperone function while it passes through the gut.(3,4) A dot blot assay using the MAbs to detect H. pylori AhpC in stool samples from humans was developed, and the results were compared to those obtained with a commonly used commercial kit. Good agreement was obtained (kappa coefficients were 0.81–0.92). In immunodot blotting for either bacteria or stool samples, distinction between positive and negative results was based on changes of color (from background color to brown) after the addition of substrate. Color of background is related to pigments of bacteria or stool. Interference of these pigments in immunodot blotting was reduced by agitating the dotted nitrocellulose membranes in PBS containing hydrogen peroxide. Lack of true cross-reactivity of enteric organisms including E. coli, Shig. flexneri, Sal. enteritidis, V. cholerae, Y. enterocolitica, and a slight cross-reaction of P. aeruginosa with MAbs and high concordance with the negative results from the ELISA kit indicated high specificity of the MAbs in dot blotting detection of H. pylori in stools. The study showed that detection limit of H. pylori AhpC for all MAbs were 8–20 μg/mL. The described limit for the detection of the purified 26 kDa antigen and high concordance results with positive results from the ELISA kit indicate high sensitivity of MAbs in dot blotting detection of H. pylori in stools. This assay does not have the limitations of [13C] UBT(5) and is a sensitive, specific, and convenient test for the eradication management of H. pylori. However, the dot blot assay developed is a qualitative test that does not offer any quantitative evaluations. Furthermore, it requires additional studies to establish its acceptability.
Footnotes
Acknowledgments
This study was financially supported by Tehran University of Medical Sciences. We acknowledge the invaluable support of the Danesh Pathobiology Laboratory (Tehran, Iran) during the collection of samples for this study.
Author Disclosure Statement
The authors have no competing interests to disclose.