
Abstract
The ever-increasing global demand for energy and materials has a pronounced effect on worldwide economic stability, diplomacy, and technical advancement. In response, a recent key research area in biotechnology has centered on the biological conversion of lignocellulosic biomass to simple sugars. Lignocellulosic biomass, converted to fermentable sugars via enzymatic hydrolysis of cell wall polysaccharides, can be utilized to generate a variety of downstream fuels and chemicals. Ethanol, in particular, has a high potential as transportation fuel to supplement or even replace gasoline derived from petroleum feedstocks. Biological or enzymatic hydrolysis offers the potential for low-cost, high-yield, and selective production of targeted chemicals and value-added co-products at milder operating conditions than thermochemical processes such as gasification or pyrolysis. Due to the complex nature of biomass, degrading enzymes, and their interactions, there is a substantial knowledge gap with respect to the mechanism of enzymatic hydrolysis and the relationship between biomass structure and enzymatic performance. This knowledge gap has greatly contributed to the fact that biological conversion of lignocellulosic biomass has not met the target performance and cost requirements for large-scale production and market entrance. This review highlights recent advances in analytical methods to characterize the chemical and molecular features related to the ability of biomass to resist biological deconstruction, defined as biomass recalcitrance. We also briefly discuss the application of some of these methods in a variety of studies that draw attention to relationships between biomass structure, the effectiveness of enzymatic hydrolysis and biomass recalcitrance.
Introduction
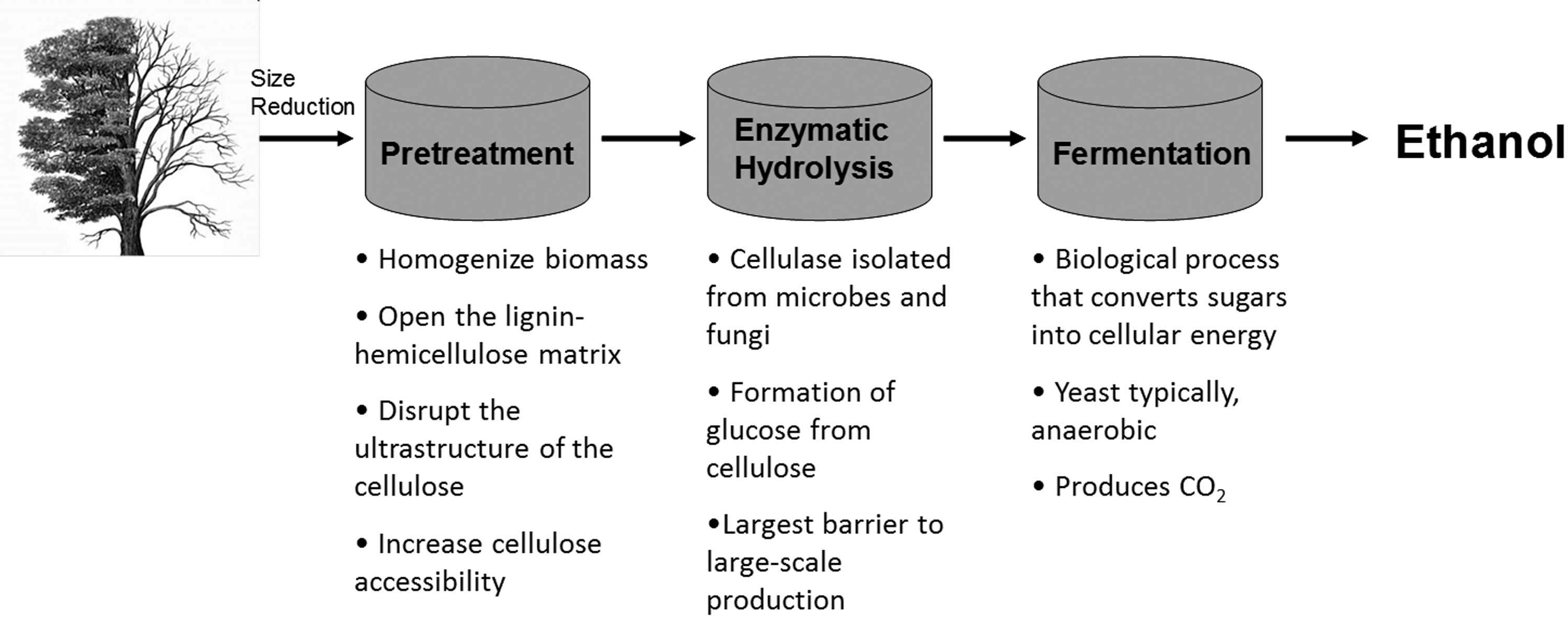
A process flow schematic describing the current biological conversion of cellulosic biomass technology.
Although supplementation and replacement of the current petroleum industry has been the subject of intense investigation for over half a century, the cost-effective production of an adequate supply of biomass-derived fuel continues to be a challenge. A major part of this effort has been the integrated “biorefinery” that would facilitate an integrated biomass conversion process to produce foods, fuels, chemicals, feeds, materials, heat, and power. 1,5 The biorefinery would effectively use the materials and energy locked within lignocellulosic biomass in a fashion analogous to modern petroleum refineries. 1,5 Currently, most biorefinery strategies rely heavily on either biological conversion of lignocellulosic biomass or thermochemical processes to produce transportation fuels as the major process flow. In order to meet global demand and produce adequate material volumes at sufficiently low cost, research must first address several issues, including but not limited to understanding plant cell wall biosynthesis and structure in the context of biomass recalcitrance; developing time-, energy-, and material-efficient conversion technologies that overcome biomass recalcitrance; and continued improvement of analytical techniques to support these tasks.
This review focuses on the latter issue, highlighting recent advances in analytical methodology to characterize the chemical and molecular features related to biomass recalcitrance. Accordingly, the scope of this review covers current studies utilizing analytical techniques to characterize biomass recalcitrance by measuring relevant substrate characteristics in a high throughput (HTP) manner that are related to cell wall biochemistry such as cell wall polymer composition, monolignol distribution, and the identity and proportion of various chemical functional groups present; that are associated with larger-scale molecular features such as specific surface area, pore structure, and accessibility; and using imaging that can indicate the cellular spatial distribution of both cell wall biochemistry and molecular features.
A critical evolutionary adaptation for plants was the biosynthesis of the cell wall, which provides structural support and protection while also facilitating the transport of water and nutrients. 6 The plant cell wall of lignocellulosic biomass contains three major biopolymers: cellulose, lignin, and hemicellulose. Cellulose is the most abundant terrestrial source of carbon and is found in the cell wall as both crystalline and amorphous morphologies consisting of linear polysaccharides composed of β-(1→4) linked D-glucopyranosyl units. 7 Hemicellulose, on the other hand, is a broad class of polysaccharide that includes several branched heteropolymers composed of a variety of 5- and 6-carbon sugars. 6 Lignin is constructed of hydroxycinnamyl monomers with various degrees of methoxylation packed into a complex, racemic, cross-linked, and highly heterogeneous aromatic macromolecule. 8 The unique chemical and physical properties of the plant cell wall can be, in part, attributed to the highly heterogeneous, multi-component nano- and microstructure of the plant cell wall. This cell wall microstructure is believed to exist as a lignin and hemicellulose matrix encapsulating and supporting cellulose microfibrils packed into bundles (Fig. 2). Also, ferulated hemicelluloses are considered potential sites for covalent cross-linking between carbohydrates and lignin, known as lignin-carbohydrate complexes. These structures make the cell wall very useful to a plant as a structural and defensive element, but consequently make it difficult and expensive to deconstruct biologically—a property known as biomass recalcitrance. Many researchers view biomass recalcitrance as the major barrier to the significant reduction in capital and processing cost for large-scale biological conversion of lignocellulosic biomass. 9 –11 Accordingly, improvements in current biomass processing and conversion technologies will require a better understanding of the mechanisms of biomass recalcitrance and its relationship to cell wall structure.
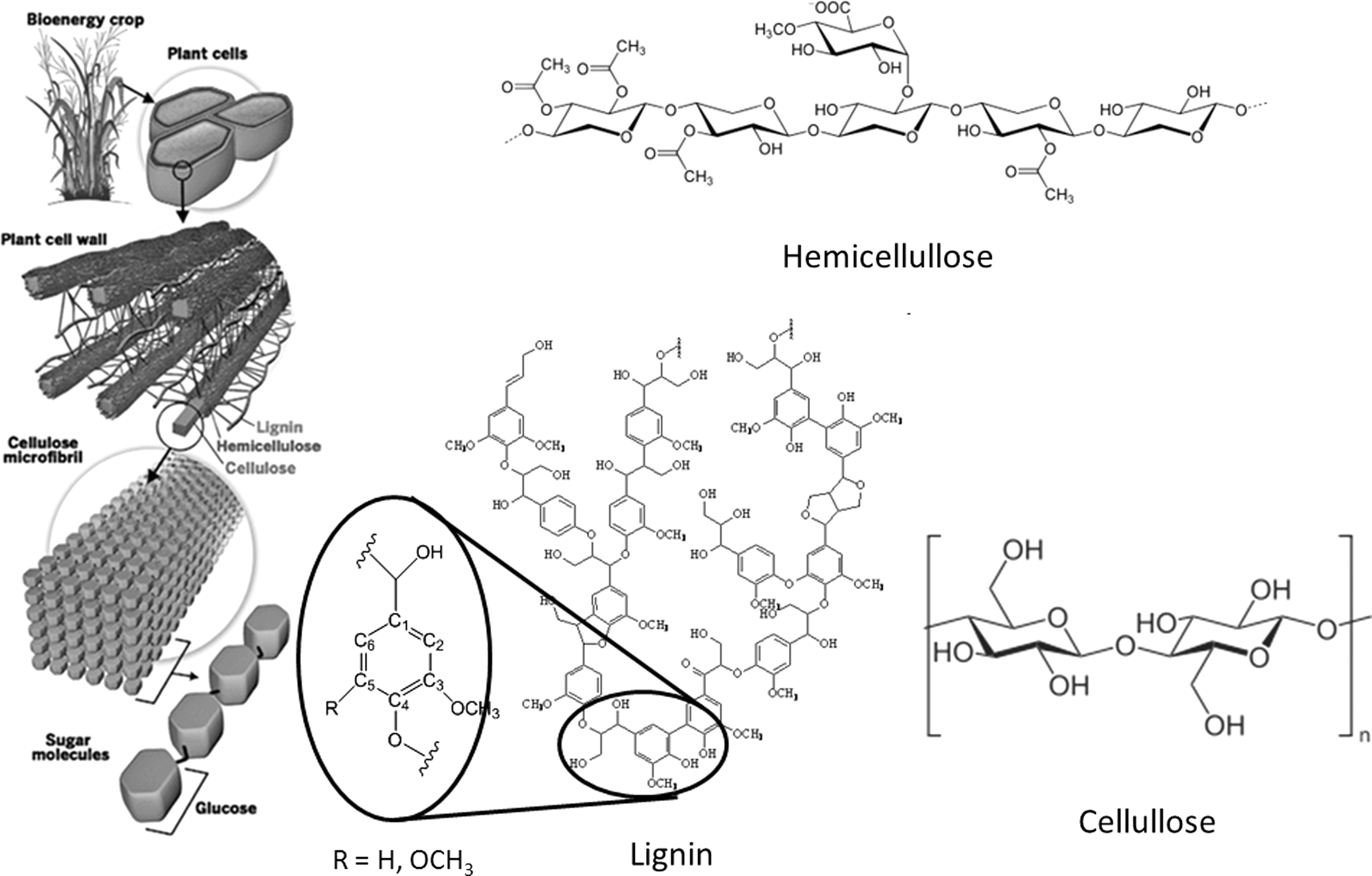
A representation of the plant cell wall and the major biopolymers in the plant cell wall where the lignin structure represents characteristic subunits in lignin and the hemicellulose represents xylan, the main hemicellulose macromolecule. The three main lignin-related monomers are defined as p-hydroxyphenyl (H) units with no methoxy groups, syringyl (S) units with methoxy groups at the C3 and C5 positions, and guaiacyl (G) units with a methoxy group at the C3 position. (Sources: Fengel F, Wegener G. Wood: Chemistry, Ultrastructure, Reaction. New York, NY: Walter de Gruyter, 1984; Lawrence Berkeley National Laboratory. Lignocellulosic Plant Cell Wall. (2010) Available at:
Origins and Mechanisms of Biomass Recalcitrance
Structural sugars, stored as cell wall polysaccharides, are a common energy source for a range of animals, fungi, and microorganisms. As a response, millions of years of evolution have honed the chemical and molecular features of the plant cell wall and its microstructure to defend those sugars. Understanding the origins and mechanisms of biomass recalcitrance is vital to advancing large-scale biological lignocellulosic biomass conversion. For example, enzymatic performance on lignocellulosic biomass has been related to the potential effects of various cell wall substrate characteristics, such as cellulose crystallinity, cellulose degree of polymerization (DP), cell wall specific surface area, lignin and hemicellulose content, and cell wall polymer monomer distribution. 9,12,13 Many of these properties affect the accessibility of the polysaccharides to enzymes. A recent review by Yang et al. covers many of the proposed properties that contribute to cell wall recalcitrance and provides perspective for improving substrates for enzymatic hydrolysis. 9
In an effort to assess directly the effects of these cell wall substrate characteristics, years of research have focused on modifying substrate characteristics and correlating substrate alterations to changes in biomass recalcitrance and sugar yields. 11,14 –17 Much of the literature, however, reports conflicting trends on the individual effects of many substrate characteristics considered important to biological deconstruction. 9,12 –15
This lack of consensus can be attributed to several issues. First, the methods used to alter specific cell wall substrate characteristics not only target plant cell wall characteristics of interest, but also invariably change a variety of other substrate properties. For example, the use of hydrothermal pretreatments has been utilized to facilitate hemicellulose removal and lignin redistribution; however, recent studies suggest the observed increases in enzymatic sugar yields and reduction in biomass recalcitrance inherent to the native material are mainly due to increases in cellulose accessibility and changes in cell wall pore structure. 15 –21 Conversely, it has been shown that these treatments also modify cellulose DP and crystallinity, which in the absence of accessibility measurements could lead to erroneous conclusions about the effect of cellulose DP and crystallinity on recalcitrance. 16 Secondly, this confusion can be partly attributed to the complexity of the plant cell wall and the fact that deconvolution of the effects of substrate properties on enzymatic hydrolysis require a comprehensive representation of the cell wall. Accomplishing this requires data collection on an array of features at multiple length scales (nm-mm)—a task few single laboratories have the capabilities to carry out. Moreover, almost all studies designed to analyze the effect of cell wall substrate characteristics on biomass recalcitrance insist on treating these relationships as simple first-order linear correlations, when more than likely a change in one characteristic will change the effect another characteristic has on biomass recalcitrance, and so forth in a recursive fashion. Many studies acknowledge this complexity, and in an effort to circumvent it focus on the effect of cell wall substrate characteristics of isolated cell wall components and their mixtures. This, however, ignores the significant relationship between cell wall component organization within intact biomass and the effects of the 3-dimensional (3D) cell wall microstructure has on biomass recalcitrance.
Tools for Biomass Characterization
To maximize the benefit of biomass as a material and energy feedstock, the ability to conduct reliable material characterization at multiple length scales, targeting specific recalcitrance-related properties is essential. Biomass can be problematic for many characterization techniques because it is a soft, hydrophilic, heterogeneous, multi-component, polymeric, and non-conducting material. However, in recent years, the development of techniques geared toward such “soft materials” has greatly expanded. Here, we discuss many of these techniques and their specific applications to biomass and characterization of biomass recalcitrance.
HIGH THROUGHPUT ANALYSIS
Many recent approaches to bypassing the limitations of biological conversion of lignocellulosic biomass have focused on altering plant substrate characteristics via genetic manipulation to generate a cell wall more amenable to conversion while continuing to develop improved pretreatments and microbial/enzymatic conversion technologies for higher-yield, lower-cost sugar release. 3,22 –30 The former part of this strategy entails searching for and targeting specific cell wall genotypes and phenotypes linked to overall sugar yields from biological deconstruction. 11,26 A recent study has shown the efficacy of this strategy by demonstrating increased sugar yields from enzymatic hydrolysis and fermentability to ethanol by simultaneous saccharification and fermentation with yeast and Clostridium thermocellum, achieved by down-regulating the caffeic acid O-methyl transferase gene in a viable switchgrass mutant. 25 Though in this case successful, the strategy suffers from a significant lack of information pertaining to which genes to alter and how those manipulations may not only alter recalcitrance, but also other cell wall properties critical to plant development and function. An in-depth understanding of the substrate characteristics critical to biomass recalcitrance and the related biosynthetic pathways is essential for identifying recalcitrance-related genes. Other tools, such as bioinformatics, association mapping, and quantitative trait locus (QTL) analysis, can also be utilized, although many of these methods require large sample sizes. 31,32 Furthermore, due to the inherent uncertainty associated with the genetic engineering of complex biological systems such as plants, screening a large number of mutant plants is typically required. HTP analytical techniques to characterize biomass recalcitrance and recalcitrance-related substrate characteristics are vital to any biomass deconstruction enhancement strategy involving genetic manipulation. This, along with significant improvements in usability, reliability, and cost of modern robotics has stimulated rapid growth in the field of laboratory automation for HTP screening in biomass-related biotechnology. 33
HTP recalcitrance screening
The most effective screen for biomass recalcitrance relies on sugar yields resulting from chemical pretreatment and/or enzymatic hydrolysis. These treatments and conventional sugar release assays typically require considerable time and manpower and are done on small bench-scale equipment (∼10–200 mL). 34 Berlin et al. developed an HTP assay to evaluate enzymatic hydrolysis of lignocellulosic substrates. 35 Later, in an effort to handle large sets of small-scale biomass samples for recalcitrance screening, researchers at National Renewable Energy Laboratory (NREL) reported the use of a novel 96-well multi-plate reactor for the comparative analysis of biomass recalcitrance by microscale hydrothermal pretreatment and enzymatic hydrolysis. 33,34,36 In this system, after automated sample loading, wells are sealed and individual 96-well plates are stacked, accounting for the multi-plate configuration. This stack can then be subjected to a variety of pretreatment conditions (biomass loading, pH, temperature, pressure, time) in a pressure reactor; this enables not only the investigation of biomass recalcitrance but also offers the possibility to optimize hydrothermal pretreatment conditions. 33 Similarly, buffer and a variety of enzymatic cocktails can be added to the 96-well multi-plate setup after pretreatment, not only to assess biomass recalcitrance but also to optimize enzymatic formulations. 33,34,37 NREL's work and similar research at Michigan State University have as well shown the viability of an HTP platform for genetically engineering energy crops, reducing pretreatment severity, and minimizing enzyme loading. 38
HTP pyrolysis molecular beam mass spectrometry (py-MBMS)
The HTP chemical analysis of biomass has included Fourier-transform infrared spectroscopy (FT-IR), near infrared spectroscopy (NIR), nuclear magnetic resonance (NMR), mass spectrometry (MS), and monoclonal antibody microarrays, which have binding specificities for cell-wall components. 39 –47 However, these techniques produce complex data sets that are challenging to interpret on large sample sets. Chemical fingerprinting of a range of materials can be effectively accomplished via the mass spectra of pyrolyzed samples. 48 –50 Analytical pyrolysis has long been used to analyze metabolite and cellular structure in a variety of complex biological systems, while more relevant examples demonstrate pyrolysis coupled with gas chromatography–mass spectrometry (GC-MS) to determine lignin content in biomass. 48 Recently, an NREL system has been used to analyze thousands of samples using py-MBMS, including rapid analysis of the chemical composition of agricultural fibers, quantitative trail locus (QTL) analysis of loblolly pine for cell wall compositional phenotypes, and investigation of within-tree lignin content variation. 48,51,52 This analysis relies on the fact that the major biopolymers in the plant cell wall depolymerize and degrade differently and in specific pathways during pyrolysis, resulting in a distinct fragmentation pattern upon MS analysis. Within these fragmentation patterns, prominent peaks can be associated with the presence of cellulose, xylan, lignin, or even syringyl (S) and guaiacyl (G) monolignol units. Typically, standards are then utilized to calculate the absolute concentration of these cell wall components. The decomposition pathways and resulting mass spectra from py-MBMS analysis can also be particularly complex; however, an attractive feature of py-MBMS, particularly with respect to HTP screening, is that large sample analysis can be accomplished by principal component analysis (PCA) of the mass fragmentation spectra. PCA can be used to group samples based on their chemical similarities and differences. For instance, in an effort to distinguish between native coconut, palm, kenaf, and flax samples, PCA of py-MBMS data was exploited to categorize various agricultural fibers treated in different ways based on the spatial grouping in a principal component plot. 53
A particularly interesting study that utilized both HTP recalcitrance screening and py-MBMS was conducted by Studer et al. 32 They analyzed 1,100 individual, undomesticated Populus trichocarpa trees in an effort to identify extreme phenotypes and correlate lignin content and the ratio of syringyl and guaiacyl units (S/G ratio) to total sugar release through enzymatic hydrolysis after hot-water pretreatment. A set of 47 extreme phenotypes was found and correlated to sugar release; among the 47 samples, sugar release profiles varied widely, with a maximum yield of ∼92%. 32 The data indicated that samples with an S/G ratio<2.0 showed a strong negative correlation between total sugar release and lignin content. However, as the S/G ratio increased above 2.0, the correlation between total sugar release and lignin content became significantly weaker. It was also determined that glucose release seemed to depend on both lignin content and S/G ratio, whereas xylose release only corresponded to S/G ratio. 32
Accordingly, we argue that lignin with a low S/G ratio has a higher likelihood of containing lower fractions of β-O-4 ether linkages and an increased number of condensed aromatic bonds, conversely generating a more fractal 3D structure. This more bulky and sterically hindering lignin reduces enzyme accessibility and deconstruction capability. Furthermore, Samuel et al. characterized ball-milled lignin from Alamo switchgrass (Panicum virgatum) before and after dilute acid pretreatment and reported the significant propensity of lignin to repolymerize during severe hydrothermal events. 54 As a result, biomass with a low S/G ratio becomes even more condensed and cross-linked after hydrothermal pretreatment, producing even more significant steric effects that may further limit enzyme accessibility and deconstruction.
DOWNSCALED BIOMASS COMPOSITIONAL ANALYSIS
Within a comparative recalcitrance screen, enzymatic sugar yields cannot be fully assessed without the concurrent compositional analysis of the starting material. A standard method for determining structural carbohydrate content in biomass uses a two-stage acid hydrolysis to digest cell wall polysaccharides into a distribution of monosaccharides. The structural carbohydrate constituents of the biomass are then determined by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD). This procedure is time consuming and requires ∼100 mg of biomass to produce sufficiently accurate results. As described above, techniques such as py-MBMS attempt to resolve the shortcomings of wet chemistry compositional analysis; however, these methods are limited with respect to chemical specificity and typically require construction of models created from large data sets of compositional results from standard methods for the interpretation of complex spectral information. 33 Based on NREL's HTP recalcitrance screening platform, Selig et al. developed a similar 96-well multi-plate setup for rapid processing of small-scale biomass samples (∼10–20 mg) by a two-stage acid hydrolysis protocol. 55 The resulting distribution of glucose and xylose was analyzed by automated glucose oxidase and xylose dehydrogenase coupled assays. DeMartini et al. accomplished downscaled biomass compositional analysis using an automated biomass-dispensing hopper and readily available liquid chromatography vials as micro-reaction vessels; digestion of cell wall polysaccharides was again completed through a two-stage acid hydrolysis with monosaccharide distribution detected via HPAEC-PAD. 56 In the latter example, approximate Klason lignin contents can be extracted from the dried acid-insoluble material remaining in the chromatography vials.
Analysis of the Chemical Features of Recalcitrance
The biochemical composition of the plant cell wall and its constituents is known to affect the efficiency of hydrothermal pretreatment, the amount and distribution of pretreatment by-products typically associated with hydrolysis and fermentation inhibition, the interactions between biomass and cellulolytic enzymes, and the rate and yield of sugars released as a result of enzymatic deconstruction. 9,11,57 The biochemical composition of the plant cell wall can also vary in response to an array of genotypic, phenotypic, and environmental growth factors inherent to the biomass source, the stage of plant development, and even the organ or tissue type from which the sample is harvested. In an effort to understand more clearly the mechanisms of biomass recalcitrance and their relationship with cell wall structure, it is critical to have methods to analyze plant cell wall chemical features.
NREL has established and standardized many of the common biomass laboratory analytical procedures for specifically targeting the chemical composition of raw biomass feedstocks and the various sample forms produced during conversion. 58 Many of these methods have American Society for Testing and Materials (ASTM) and Technical Association of the Pulp and Paper Industry (TAPPI) versions and include: the analysis of structural carbohydrate monosaccharide distribution by two-stage acid-hydrolysis and high-performance liquid chromatography (HPLC) HPAEC-PAD, determination of lignin content as defined by acid-insoluble (residual solids after two stage acid-hydrolysis measured by mass) and acid-soluble (measured by ultraviolet-visible spectroscopy) varieties, along with methods for the measurement of protein, ash, and extractive contents. 59 Numerous other wet chemistry procedures can be used to investigate cell wall composition. Hatfield et al. revisited the acetyl bromide assay developed in the 1960s to provide a rapid and sensitive method for quantifying lignin in woody plant species by spectrophotometry, evaluating key modifications to the original method. 60 Chang et al. extended the utility of the acetyl bromide assay by developing a rapid microscale determination of lignin content for HTP analysis. 61
The biochemistry of the plant cell wall extends to smaller length scales than composition; the presence and amount of individual chemical moieties and functional groups on its component constituents also have a large effect on pretreatment, enzymatic hydrolysis, and biomass recalcitrance. Analyzing these specific functionalities typically requires cell wall component isolation to deconvolute their origin, primarily due to chemical feature commonality between various constituents of the plant cell wall. These functionalities or chemical moieties can be further investigated by wet chemistry methods such as titration to determine carbonyl, carboxyl, methoxy, phenolic, ethylenic, or sulfonate group content; however more recent focus has relied on spectroscopy. 8
FT-IR has been extensively applied in plant cell wall analysis for comparing differences between samples due to cell wall developmental and compositional changes. 62 The resulting FT-IR spectrum is a complex fingerprint representing adsorption bands that correspond to the frequencies of vibrations between the bonds of the chemical functional groups comprising the cell wall. It can be difficult to quantify this data or even correctly attribute the functionality to the proper chemical moiety or component within the cell wall without proper models.
NMR, on the other hand, provides a high level of detailed chemical and structural information that is especially useful in the analysis of isolated lignin. 63 One-dimensional (1D) 1 H NMR can quantify a number of important chemical features in isolated or acetylated lignin. Acetylating lignin generally improves solubility and the resulting resolution helping to identify key functionality, including carboxylic acids, aldehydes, phenolic hydroxyls, β–5 phenolic hydroxyls, syringyl C5 phenolic hydroxyls, aromatic protons, and aliphatic protons. 64 –67 1D 13C NMR of isolated lignin is also particularly useful in the analysis and quantification of aryl ether, condensed and uncondensed aromatic and aliphatic carbons, and is ultimately capable of determining monolignol distributions. 54,68,69 Two-dimensional (2D) 1H-13C NMR experiments, which are not quantitative but help resolve a variety of overlapping spectral features, are even more useful, providing information about a wide array of chemical moieties present. 54 Analysis of other NMR nuclei based on derivatized lignin can also provide quantitative data on the concentration of a variety of other functionalities, such as a 19 F NMR procedure to investigate carbonyl functional groups or 31 P NMR to measure the combined ortho– and para–quinone contents or to determine terminal chain hydroxyl distribution and content. 70 –72 Samuel et al. characterized ball-milled lignin isolated from Alamo switchgrass (Panicum virgatum) by 13C and 31 P NMR spectroscopy before and after hydrothermal pretreatment, and was able to determine that the major lignin inter-unit linkage was β-O-4 ether bonds and that minor amounts of phenylcoumarin, resinol, and spirodienone were present. 54 They also found that acid-catalyzed hydrothermal pretreatment caused chain scission in lignin, primarily at β-O-4 ether bonds, while decreasing the S/G ratio from 0.80 to 0.53. 54 Most importantly, however, they showed that a change in the lignin molecular weight, as determined by gel permeation chromatography (GPC), did not correspond to the aliphatic chain and monolignol inter-unit linkage degradation, which was observed spectrally. This suggests that condensation reactions also occur during pretreatment, providing a possible explanation for why lignin persists and aggregation is so prevalent under harsh pretreatment conditions. 54
Hemicellulose and pectin, including various arabinogalactans, glucomannans, galactoglucomannans, and xyloglucans, can be extracted from the cell wall using procedures typically involving lignin removal via acidic sodium chlorite holocellulose pulping followed by a combination of organic and alkaline extractions. 73,74 In this case, either polymeric or hydrolyzed monosaccharides can be analyzed via 1D 1H and 13C NMR in addition to 2D 1H-1H and 1H-13C correlation NMR. 73,74
FLOW-THROUGH PRETREATMENTS
Heterogeneous complexity and low solubility are two of the most challenging biomass characteristics that hinder easy chemical analysis. In recent unpublished work, we demonstrated that flow-through pretreatment is an ideal method for systematically deconstructing the cell wall for analytical purposes in a relatively facile, controlled, and repeatable fashion, effectively reducing sample complexity while increasing analyte solubility. Hot water and dilute acid flow-through pretreatment systems have been developed as alternatives to conventional batch systems, offering advantages such as more digestible lignocellulosic substrates, near-theoretical hemicellulose recovery, greater lignin removal, and less fermentation inhibitor generation. 29,75,76 The flow-through pretreatment strategy depicted in Figure 3 involves passing a pretreatment media with a specific pH and temperature profile in a continuous fashion through a reactor containing biomass for defined residence times. Despite these benefits, large-scale commercial implementation of flow-through pretreatment is not feasible, mainly because large amounts of water are required and the cost associated with energy use for pretreatment and sugar recovery is excessive. There are also challenges related to the scale-up of the flow-through configuration. Nonetheless, flow-through pretreatments can provide unique insights into pretreatment chemistry; Wyman et al. clearly demonstrated this and the usefulness of flow-through pretreatment as a method to conduct the controlled fractionation of the plant cell wall, in which less recalcitrant chemical moieties elute from the reactor at a much quicker rate than more recalcitrant structures. 77 –80 Consequently, flow-through pretreatment can be viewed as an additional analytical tool to probe the structure of the plant cell wall, biomass recalcitrance, and the mechanisms of cell wall component release during more conventional batch pretreatments.

Schematic of the flow-through reactor system.
Liu et al. studied the effect of flow rate on the fate of hemicellulose and lignin during hot water pretreatment of corn stover at various temperatures and found that removal of xylan and lignin were nearly linearly related. 79 This observation seemed to suggest that the eluting lignin and hemicellulose fragments were linked in some manner, possibly via covalent lignin-carbohydrate linkages. The authors hypothesized that in a batch system these fragments continue to react during pretreatment, forming insoluble species that precipitate out of solution and cause lower lignin removal and higher lignin condensation, aggregation, or droplet formation. 79
SEQUENTIAL EXTRACTIONS
Sequential extraction, in which biomass is exposed to increasingly harsh reagents to solubilize different cell wall components, is another method used to achieve controlled fractionation of the plant cell wall. The use of these reagents to target specific cell wall components is particularly useful in the analysis of recalcitrance because their relative ability to cause deconstruction directly corresponds to the strength of binding for the particular chemical moieties eluting from the cell wall. A sequential extraction procedure for biomass was developed at the Complex Carbohydrate Research Center (CCRC) at the University of Georgia; it utilizes an oxalate, carbonate, 1.0 M KOH, 4.0 M KOH, chlorite, and post-chlorite 4.0 M KOH extraction sequence (Fig. 4). 81 Mild reagents such as oxalate and carbonate tend to extract arabinogalactans and pectic cell wall components. The harsher alkaline reagents release more recalcitrant structures like xylans and xyloglucans, while chlorite reagents target even more tightly bound lignin constituents. The extractions, in conjunction with a variety of analytical techniques such as NMR or MS on either the extracts or residue solids, can effectively determine which chemical moieties are removed and their relative recalcitrance.
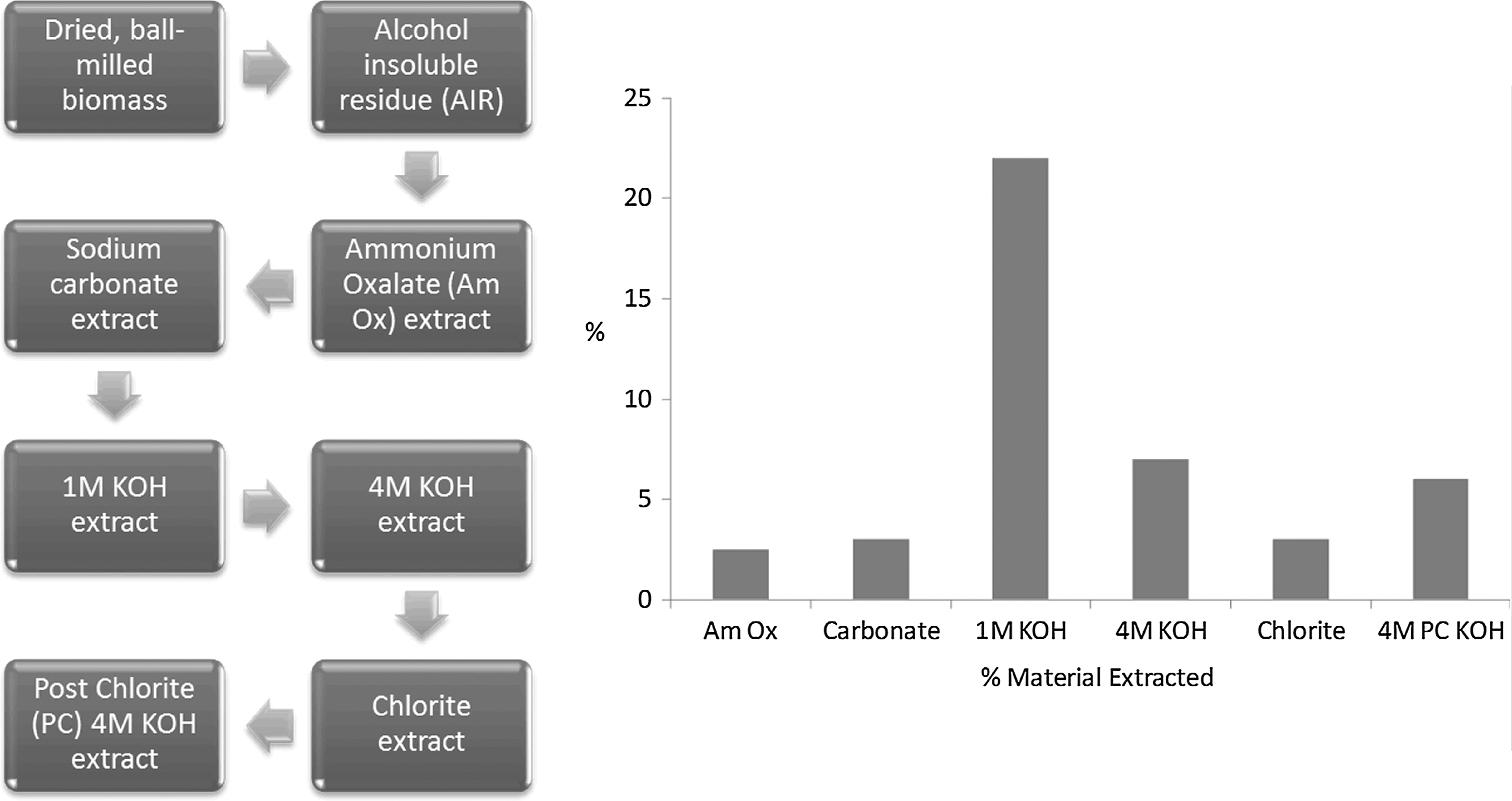
The sequential extraction procedure developed at the CCRC.
DeMartini et al. performed this sequential extraction method on hydrothermally pretreated Populus under increasing residence times. 81 The extracts and residual solids were analyzed by plant glycan-directed monoclonal antibody assays and immunolabeling, respectively. They determined that the sequence of structural changes that occurs in plant cell walls during pretreatment begins with disruption of lignin-polysaccharide interactions and significant removal of pectins and arabinogalactans. As pretreatment progresses, the process of hydrothermal cell wall disruption is then dominated by removal of xylans and xyloglucans. 81 A similar study investigating alterations in lignin structure with antisense down-regulation of p-coumarate 3-hydroxylase (C3H) or hydroxycinnamoyl-CoA:shikimate hydroxycinnamoyltransferase (HCT) expression in alfalfa (Medicago sativa) used a simplified sequential extraction procedure of 0.1 M KOH followed by 2.0 M KOH. 82 In this case, the residues after extractions were analyzed by solid state NMR and GPC, while the hydroxyphenyl (H), G, and S monolignol content in the extracts were characterized by thioacidolysis/GC-MS. The results seem to indicate that C3H and HCT down-regulation increased lignin extractability, due in part to a change in the monolignol distribution towards increased H monomer and a decrease in the molecular weight. 82 The study further demonstrates the efficacy of strategies to reduce biomass recalcitrance by genetic modification by showing that changes to lignin biosynthesis can change lignin molecular weight and chemical processability.
GLYCOME PROFILING
Because of the complexity and heterogeneity of the plant cell wall, chemical characterization by spectroscopy can be at times intricate and complicated. The numerous, distinct chemical moieties are typically exhibited as a complex, overlapping spectral signature. Therefore, several groups have begun to utilize monoclonal antibodies (mAbs) with specificities for glycan cell-wall components and carbohydrate-binding modules (CBMs) from microbial hydrolases for effective monitoring of glycan-related epitopes of cellulose, hemicellulose, and pectin. 46,47,81,83 Early work by Moller et al. focused on the semi-quantitative HTP mapping of glycans from multiple organs or tissues by microarrays. 46 In this case, 18 mAbs and two CBMs were used to monitor 13 glycan epitopes of various sequential extracts of Arabidopsis thaliana mutants. A later, highly relevant study used HTP microarray glycome profiling of wheat straw during hydrothermal pretreatment. 47 A much larger and diverse antibody toolkit which comprised of more than 155 antibodies was used by DeMartini et al. to yield a more complete depiction of alterations occurring in the cell wall during hydrothermal pretreatment, including epitopes for pectins, galactomannans, arabinogalactans, xylans, fucosylated and non-fucosylated xyloglucans, and lignin-associated glycans. 81 The power of glycome profiling is further extended by immunolabeling, which can provide information detailing the spatial distribution of carbohydrate epitopes in intact biomass in situ. Ultimately, using the larger antibody library, DeMartini et al. determined that the integration and association of lignin and polysaccharides within the cell wall play a more critical role in biomass recalcitrance than lignin content alone. 81
LIQUID-STATE NMR SPECTROSCOPY
Destructive techniques such as py-MBMS and non-destructive techniques such as NIR and FT-IR have been applied to plant cell wall chemical analysis. 48,53,62 However, few techniques can provide the type of detailed chemical and structural information, spectral resolution, and sensitivity as solution NMR. The intact cell wall itself is inherently insoluble in almost all solvent systems, so traditional solution NMR methods have relied on time-consuming extraction and purification techniques to characterize the plant cell wall. However, there is a high probability that these extraction and purification techniques alter the innate chemical structure of the isolated cell wall components. To avoid cell wall component isolation while facilitating the characterization of non-derivatized biomass, Kim et al. developed a preparation that simply involves moderate size reduction by ball-milling and swelling of the cell wall with DMSO-d6. 84,85 The swollen cell wall material has enough molecular mobility to produce spectral data similar to high resolution liquid-state NMR spectra.
Ionic liquids have become popular as green solvents uniquely suited for the dissolution and processing of biomass and plant cell wall components. Swatloski et al. has shown that ionic liquids are good solvents for cellulose and biomass, due in part to the chloride anion that helps dissolve cellulose by disrupting the strong hydrogen bonding network. 86 In a study examining the application of select ionic liquids as aprotic green solvents for lignin, Jiang et al. demonstrated that up to 20 wt% solutions of lignin could be generated in 1-hexyl-3-methylimidazolium trifluoromethanesulfonate, 1,3-dimethylimidazolium methylsulfate, and 1-butyl-3-methylimidazolium methylsulfate. The high solubility facilitated effective 1D 13C NMR analysis, and resulting spectra indicated that lignin and lignin model compounds displayed only a slight up-field change in chemical shift with respect to the signature obtained with DMSO as the solvent. 87 A variety of other citations also pertain to the dissolution of polysaccharides in ionic liquid solvent systems to analyze their structure by 13C NMR. 88 Nevertheless, chemical analysis by this 1D method, especially for intact cell wall material, is difficult due to the complex and overlapping nature of the spectra, obscuring signatures from the ionic liquids/solvents, and to long relaxation times, resulting in long acquisition times and yielding low signal-to-noise (S/N) ratio.
Yelle et al. demonstrated the first use of a liquid solvent system, 1-methyl imidazole-d6/DMSO-d6, to swell/dissolve lignocellulosics while also utilizing 2D 13C-1H heteronuclear single quantum coherence (HSQC) NMR experiments to shorten acquisition times, improve S/N, and increase spectral resolution over two dimensions. 89 This study effectively analyzed cell wall chemistry in intact biomass from pine, quaking aspen, and kenaf, identifying various lignin subunits including S and G monolignol units, resinol, phenylcoumaran, β-aryl ether monolignol linkages, and polysaccharide units such as acetylated xylopyranosides. More recently, Ragauskas et al. published a series of manuscripts using a perdeuterated ionic liquid —pyridinium chloride-d6/DMSO-d6—as a cheap and easily synthesized alternative solvent system for dissolving Wiley milled biomass. 87,90 –92 Foston et al. used this solvent system and 2D NMR in part to analyze the underlying phenotypic, biochemical, and morphological properties associated with the reduced recalcitrance observed in tension stress-induced reaction wood of Populus. This technique indicated a qualitative decrease in the proportion of β-aryl ether linkages and a slight increase in resinol sub-structures when comparing tension wood to normal wood. Other key observations from the spectral data included significant changes in the C2-acetylated xylopyranoside:C3-acetylated xylopyranoside ratio and distribution of para-hydroxybenzoate (PB) to S to G monolignol units. 90
SEQUENCING OF XYLO-OLIGOSACCHARIDES BY MASS AND NMR SPECTROSCOPY
A major polysaccharide constituent in the cell wall of grasses is arabinoxylans, which consist mainly of a backbone of β-(1→4) linked D-xylopyranosyl residues with a variety of arabinose-containing side chain units originating from the xylopyranosyl 2- and 3-positions. 93 The resulting polysaccharide monomer sequence not only affects cell wall chemistry but also influences molecular features such as the degree of branching and spatial arrangement of hemicellulose, which has been shown to correlate with altered cell wall properties. 93 Although no study has shown a direct link between substrate characteristics such as hemicellulose monosaccharide unit distribution and sequence, the linkage position for each glycosidic bond, and the presence and location of each branch point in hemicellulose with biomass recalcitrance, it is clear that these factors will alter the 3D cell wall microstructure and subsequent cellulose accessibility before and after pretreatment.
Mazumder et al. demonstrated the detailed structural analysis of these arabinoxylans by utilizing a 1.0 M KOH-extraction followed by treatment with a Trichoderma viride endo-(1→4)-xylanase for controlled fragmentation of the extracted arabinoxylans to generate oligosaccharides. 93 These oligosaccharides were further purified by size-exclusion chromatography, producing five fractions from alcohol-extracted switchgrass (Panicum virgatum var Alamo). The monosaccharide composition and glycosidic linkage distribution of these arabinoxylans fragments were analyzed using a combination of HPAEC-PAD and GC–MS of monosaccharides and partially methylated alditol acetate derivatives generated by acid-catalyzed depolymerization and/or per-O-methylation, hydrolysis, reduction, and acetylation of each fraction. 93 This analysis indicated that Alamo switchgrass arabinoxylan purified fractions 2, 3, and 4 contained arabinose/xylose ratios of 6.7, 2.1, and 3.9, respectively, while methylation analysis demonstrated that arabinoxylan purified fractions 2 and 4 consist of (1→4)-linked xylopyranosyl residues with a single arabinofuranosyl side chain coupled to the 3-postion of an internal xylose unit.
The molecular weights of the purified oligosaccharide fractions were probed using matrix-assisted laser desorption/ionization MS (MALDI-TOF MS), and glycosyl sequences and branching topologies were determined by multiple-stage electrospray-ionization MS (ESI MSn) and 1H NMR spectroscopy, clearly showing that switchgrass arabinoxylan purified fractions 2, 3, and 4 contained arabinoxylans with a backbone made of linear β-D-(1→4)-xylopyranosy units, with α-L-arabinofuranosyl-(1→and α-L-arabinofuranosyl-(1→2)-α-L-arabinofuranosyl-(1→side chains at the 3-postion of internal xylose units. 93
Analysis of the Molecular Features of Recalcitrance
Biomass recalcitrance is a complex, multi-variant, multi-scale problem; while recalcitrance-related substrate characteristics can extend to length-scales beyond cell wall chemistry. 9,12,14,15 It is therefore critical to characterize and have methodologies to target larger-scale molecular features such as specific surface area, pore structure and accessibility, cell wall compositional spatial distribution, and cellulose ultrastructure to gain a better understanding of the mechanisms of biomass recalcitrance and the relationship with cell wall structure. For instance, efficient enzymatic hydrolysis of lignocellulose has been directly related to cellulase enzyme activity and the potential effect of ultrastructural characteristics of cellulose, including crystallinity, crystal lattice structure, crystallite dimensions, and DP. 12
The role of cellulose DP on enzymatic hydrolysis is not clearly defined in the literature for many of the reasons discussed in the introduction. 12,14 In a review focused on determining changes in cellulose DP before and after various types of pretreatment, along with the effect of DP on enzymatic deconstruction of cellulose, a major conclusion indicated that shorter cellulose chains can be more readily hydrolyzed enzymatically. 94 Whereas, Del Rio et al. showed that single substrate characteristics such as fiber length, cellulose degree of polymerization, and crystallinity have little effect on a substrate's susceptibility to enzymatic hydrolysis by studying the enzymatic hydrolysis of organosolv-pretreated softwood materials. 95 However, it is reasonable to at least expect that certain exoglucanases may display higher activity with increasing number of reducing chain ends. Considering the differing opinions on the subject, accurate analysis of cellulose DP is even more critical, requiring data interpretation in the context of multiple recalcitrance-contributing characteristics.
The average molecular weight of a polydisperse distribution of polymers is typically expressed as an ordinary arithmetic mean or weighted mean denoted as the number average molar mass (Mn) and weight average molar mass (Mw), respectively. A variety of methods, often only measuring either Mn or Mw, can be used to determine the relative molecular weight of cellulose, including vapor pressure osmometry, reducing end concentration, electron microscopy, light scattering, sedimentation equilibrium, and intrinsic viscosity. 96 Moreover, solution-based techniques typically require solublization via derivatization. A commonly utilized technique generates cellulose tricarbanilate with phenyl isocyanate from isolated cellulose to facilitate dissolution in tetrahydrofuran (THF) and elution in a GPC system. 97,98 Though this method involves derivatization, specialized chromatography setups, and external standards, it is one of the few techniques that can provide not only an average molecular weight but reproduce the entire molecular weight distribution. In highly nonsymmetric, multi-modal distributions that do not fit a theoretical molecular weight model, which is the case with many natural polymers, having the entire molecular weight distribution is a powerful tool for data interpretation. In addition, once set up, automated GPC systems can process large numbers of samples.
Cellulose crystallinity is also a frequently measured parameter, because crystallinity, crystal lattice structure, and crystallite dimensions can alter material properties. A common observation in the study of cellulose degradation has shown that crystalline cellulose has a much slower rate of acid- or auto-catalyzed hydrolysis than amorphous cellulose. 99 Furthermore, Pu et al., in a study tracking the ultrastructural features of cellulose from bleached softwood kraft pulps during hydrolysis with cellulase from T. reesei, found that different types of crystalline cellulose allomorphs (cellulose Iα and cellulose Iβ), para-crystalline cellulose, and amorphous cellulose were consumed by cellulase to different extents and at different rates. 100 In an initial, rapid phase of hydrolysis para-crystalline and non-crystalline cellulose regions were more readily enzymatically deconstructed than cellulose Iα, which in turn displayed a faster rate of hydrolysis than cellulose Iβ. This suggests that not only does cellulose crystallinity affect enzymatic hydrolysis and recalcitrance, but that crystal lattice structure may also be important.
The literature frequently suggests that lower cellulose crystallinities are a key characteristic for increased sugar yields from deconstruction. This is evident in a study by Hall et al. in which phosphoric acid pretreatment of Avicel produced substrates of low crystallinity and high enzymatic digestibility. 101 However, similar to cellulose DP, the exact contribution of crystallinity with respect to the myriad of multiple recalcitrance-contributing characteristics in native biomass is uncertain and varies widely in the literature. The correlation between cellulose crystallinity and recalcitrance demands, at a minimum, analysis to help establish its relative importance.
Crystallinity in cellulose is typically evaluated as the ratio of two contrasting and distinct phases, defined as either crystalline versus amorphous; solvent inaccessible versus accessible; or ordered versus disordered domains. 7,102,103 Techniques to determine the ratio of these phases typically rely on one of three approaches: physical methods, generally measured as a function of disorder (i.e., density); chemical swelling methods, in which reagents swell cellulose by disrupting hydrogen and inter-chain bonding, penetrating the cellulose molecular structure in the amorphous region by either non-reactive means such as moisture regain or iodine sorption, or reactive means such as formylation or acid hydrolysis; or chemical non-swelling methods, which instead of disrupting the hydrogen and inter-chain bonding, by only interacting with external crystalline surfaces and detecting the proportion of cellulose crystal surfaces as well as surfaces of internal voids, capillaries, and fibril structures–i.e., N2 adsorption, chromic acid oxidation, or thallation. 102,103
Physically evaluating the relative fraction and size of crystallites in cellulose can also be accomplished through IR or Raman spectroscopy along with X-ray diffraction (XRD) and electron diffraction/microscopic methodologies. 104,105 A common XRD method to determine cellulose crystallinity from randomly oriented fibers of isolated cellulose considers the peak intensity of the 002 diffraction plane (specific for cellulose I) compared to the intensity at the same diffraction plane from that of pure amorphous or regenerated cellulose. 104 There are, however, several other methods to process XRD patterns and determine cellulose crystallinity, and, though widely used for decades, many researchers argue that this method does not provide a true crystallinity and that only fiber diffraction techniques, and the analysis of diffraction spots versus diffraction ring patterns, can generate accurate results. 105 –107 IR and Raman spectroscopy, on the other hand, generate ratios of the peak intensity at various vibrational bands assigned to crystalline and amorphous cellulose; for example, % crystallinity=I1481/(I1481+I1462), where Raman peaks at 1,481 cm−1 and 1,462 cm−1 represent crystalline and amorphous cellulose, respectively. 104 Whereas crystallinity by IR can be determined by the ratio of peak height or area at 1280 cm−1, considered a crystalline cellulose band, to a relatively constant band at 1200 cm−1; however, many of these crystallinity related assignments, both Raman and IR, vary widely in literature. 104
Another useful tool to analyze the ultrastructural features of cellulose is 13C cross polarization magic angle spinning (CP/MAS) NMR spectroscopy. 7 Because solid-state NMR is sensitive to the magnetic non-equivalences in an environment of chemically equivalent nuclei, crystalline and amorphous cellulose have been shown to generate different chemical shifts. Atalla and Vanderhart first reported the use of 13C CP/MAS NMR to study cellulose from Acetobacter, Valonia, kraft pulp, and low-DP acid-hydrolyzed cellulose, and observed variations in chemical shifts for the C4 and C6 carbon positions in the anhydroglucose unit dependent upon the cellulosic source. 108 –111 They attributed this to variations in cellulose crystallinity and crystal lattice structure. 110,111
In the 13C CP/MAS NMR spectrum of isolated cellulose, the C1, C4, and C6 signals of cellulose extend over chemical shift ranges of δ∼ 102–108, 80–92, and 57–67 ppm, respectively. 7,112 Though the appearance of each of these peaks can be used to extract ultrastructural information, the C4 peak is most commonly utilized (Fig. 5). C4 cellulose amorphous chains are represented by a fairly broad signal from δ ∼80–85 ppm, while C4 cellulose crystalline chains generate a sharper resonance from δ ∼85–92 ppm. 7 Using either a two-peak, non-linear, least-squared fit or basic peak integration of these two regions, degree of cellulose crystallinity can be determined. In an effort to estimate the relative amounts of cellulose crystallinity involving cellulose Iα and cellulose Iβ, Lennholm et al. developed a novel method utilizing a partial least-squares (PLS) model. 113 This analysis was later extended to estimate not only cellulose Iα and cellulose Iβ content, but also to deconvolute contributions of para-crystalline cellulose (described as sub-crystalline cellulose having more order than amorphous cellulose and less order than crystalline cellulose) and accessible and inaccessible fibril surfaces based on the non-linear line-fitting of six or seven resonances to the C4 peak region of adjustable shape, width, chemical shift, and relative intensity. 114,115 Subsequent studies suggested that cellulose crystallite or microfibril dimensions can be estimated using the relative intensity of C4 amorphous peaks. 116 –119 In that case, the C4 amorphous region represents the percentage of chains at fibril surfaces along a square cross-sectional cellulose microfibril model; where cellulose chains have a width of 0.55 nm, the cellulose microfibril and microfibril bundle dimensions can then be calculated. 116 –119
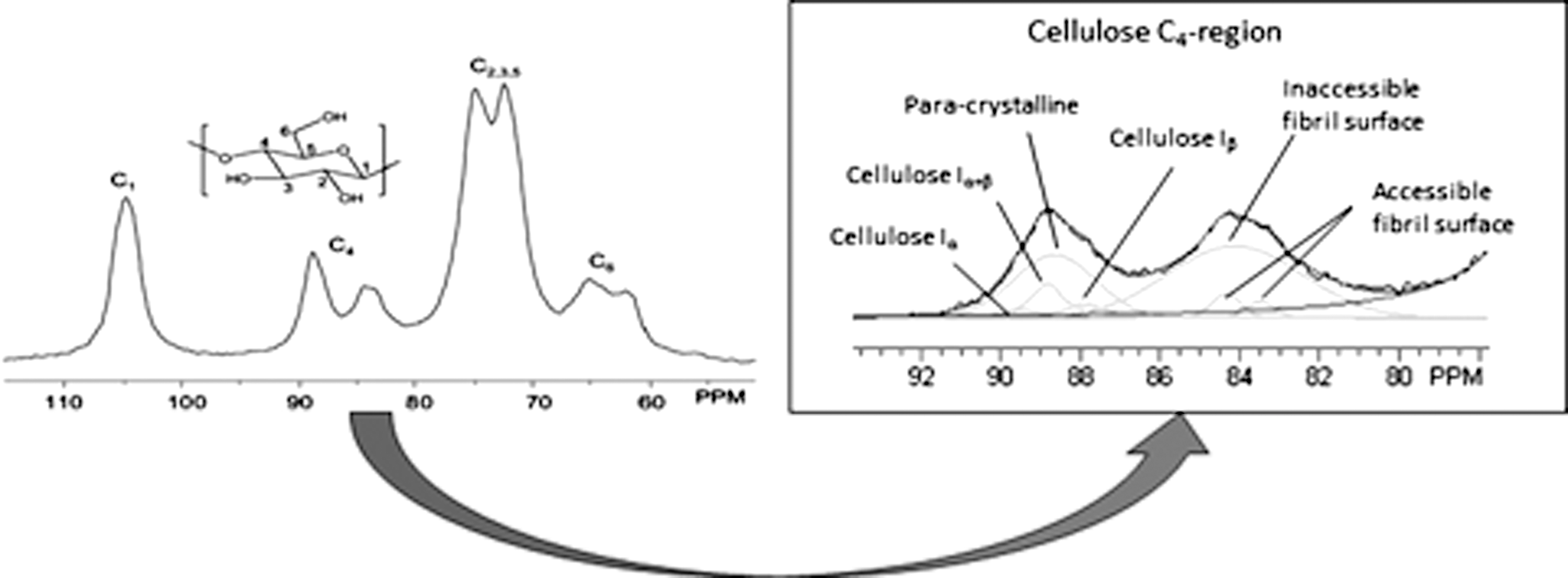
A characteristic spectrum of cellulose and its C4-carbon region (used to determined cellulose ultrastructural properties).
Using 13C CP/MAS NMR experiments on cellulose isolated from dilute acid pretreated Populus, Foston et al. determined the relative intensity of cellulosic ultrastructural components in pretreated poplar samples and how those relative intensities changed with increasing pretreatment residence time or severity. 16 13C CP/MAS NMR and non-linear line-fitting showed that the crystallinity index and cellulose crystallite dimension increased with pretreatment residence time. 16 These findings were significant because they contradicted a widely held position that a critical function of pretreatment is the de-crystallization of cellulose. The results also showed that, while the relative intensity of the other crystalline allomorphs remain relatively constant with pretreatment residence time, the relative intensity of crystalline cellulose Iα decreased and para-crystalline cellulose increased. 16 This suggests that the crystal Iα form is particularly susceptible to either degradation by acidic hydrolyzation or to thermally induced transformation to para-crystalline cellulose or other crystal polymorphs during pretreatment.
SOLID-STATE NMR SPECTROSCOPY
Solid-state NMR can be a powerful tool to analyze molecular features of the plant cell wall because it keeps the native 3D ultrastructure fully intact. An interesting application demonstrating the utility of solid-state NMR is work done attempting to determine the average spatial dimensions between major biopolymers within the plant cell wall. This was accomplished on highly 13C-enriched corn stover stems using a solid-state NMR technique referred to as 13C CP with a selective destruction of magnetization (SELDOM) filter and mixing time delay for a spin diffusion experiment. 120 The SELDOM sequence functions as a filter, localizing nuclear magnetization only on specific aromatic lignin carbons, and, as in any non-equilibrium state, this magnetization will diffuse during a mixing time delay to surrounding nuclei until equilibration is achieved. 120 According to the results of this study, the magnetization transfer pattern indicates that lignin is in close proximity to hemicellulose, followed by amorphous and finally crystalline cellulose. The rates of magnetization transfer suggest that the plant cell wall structure is intimately blended, with no major component greater than ∼2 nm from one another, and that the cellulose microfibril has a core-shell configuration with the amorphous shell being ∼1 nm thick around the crystalline core. 120
The route for enzymes to gain access to cellulose inside the cell wall is closely related to cell wall pore size and structure and, therefore, is a key determining factor in the bioconversion of lignocellulose, biomass recalcitrance, and enzymatic saccharification. 18 –20 Pore structure and accessibility can be assessed by several means: solute exclusion, a technique based on the accessibility of different sized solute molecules; differential scanning calorimetry, based on the principle that water contained inside pores has a lower freezing point than that of bulk water; a closely related technique, cryoporometry NMR; gas adsorption; or the adsorption of fluorescently tagged CBMs or polymeric dyes. 18,121 –125 However, Foston et al. utilized relaxometry NMR and a solvent probe, water, to investigate changes in the supramolecular pore structure of Populus as a result of dilute acid pretreatment. 21 Water can be found spatially localized on and within cellulosic microfibrils, existing as capillary water in the lumen or between fibers and within the lignocellulose fibril matrix. The type and strength of this association, particularly in the cell wall material, are directly related to the ultrastructural and chemical state of the lignocellulose. 126 These types of cellulose-water interactions exist in native wood pulps; however, as pretreatment is performed, material is solubilized and removed from the biomass while also altering the ultrastructural state of the cellulose. 126 By monitoring either the diffusive and/or relaxation behavior of the adsorbed water, information about changes in pore structure of lignocellulosic material can be inferred. 127,128
Foston et al. illustrated that spin-spin relaxation times of water adsorbed into pretreated biomass shifted to longer relaxation or a more mobile state, while also indicating that the relative population of water with longer relaxation times increases. 21 These changes suggest that pretreatment begins to break down and loosen the lignocellulosic ultrastructure within the biomass, increasing the average pore size. 21 Changes in 2H spin-lattice times of deuterium oxide adsorbed into pretreated biomass appear to support this, while adsorbed water diffusion coefficients, determined by pulse field gradient experiments, suggest that the pore structures were becoming more tortuous with pretreatment as well. 21
IMAGING BIOPOLYMERS
Cellular spatial distribution of chemical attributes, such as the chemical functionality distributions/biopolymer composition, and of molecular attributes, such as cellulose crystallinity or DP, further complicate efforts to understand biomass recalcitrance and its relationship to cell wall structure. Biomass milling and size reduction have been utilized in part as a method to homogenize substrate for analysis and, more relevantly, deconstruction, eliminating the effect of varying cellular, chemical, and molecular feature spatial distribution. To accommodate ease of handling, homogeneous or averaged sampling for various analytical techniques, and smaller pretreatment and enzymatic hydrolysis reactors, most laboratory biofuel research has utilized small biomass particle sizes (typically<2mm). Size reduction requires significant energy inputs, particularly for woody biomass, and therefore the use of larger particle sizes in industrial-scale processes is critical to achieve economically viable product yields.
However, little is known about the type and amount of heterogeneity that exist at these larger-length scales, and how that may affect biomass recalcitrance. The plant cell wall is typically deposited in three layers, and serves as an ideal example of how an even larger-length-scale of organization and potential chemical/molecular heterogeneity that may affect recalcitrance. The first cell wall layer to form is the middle lamella, which is generally rich in pectins and considered to be the interface that bonds adjacent plant cells together. The next layer to deposit during cell growth is the primary cell wall, generally a thin and flexible layer composed of major carbohydrates (cellulose, hemicellulose, and pectin). The secondary cell wall is the last to deposit inside the primary cell wall after the cell is fully grown. It includes carbohydrates, lignin, and a variety of other biomolecules that, depending on cellular function, modify mechanical properties and permeability. Not found in all cell types, the secondary cell wall is the predominant structure in woody biomass and may be composed of three distinct sub-layers—S1, S2, and S3–in which the directional alignment of the cellulose microfibrils differ. These cell wall layers can be effectively imaged using common transmission or scanning electron microscopy after cryotoming to preserve cellular structure during cross-sectioning. 104
Foston et al. found significant variation in lignin and cellulose deposition for tension stress-induced reaction wood from Populus with respect to normal wood. 90 The tension wood displayed enhanced sugar release profiles, in part attributed to the significant localization of lignin to cell corners and middle lamella versus secondary cell wall layers. 90 Other studies on similar tension wood substrates have also detected significant changes in the S/G ratio of lignin and cellulose microfibril crystallinity, orientation, and dimensions, depending on which secondary cell wall sub-layer was analyzed. 129 –132 These localized changes in the chemical/molecular attributes of major secondary cell wall components in tension stress-induced reaction wood seem to have a considerable effect on enzymatic hydrolysis and biomass recalcitrance. It is therefore critical in studies investigating and establishing the fundamental mechanisms of plant cell wall recalcitrance to have available methods for characterizing the cellular spatial distribution of chemical and molecular features.
ToF-SIMS and MALDI-MS
Secondary ion mass spectrometry (SIMS) uses a focused ion beam to release neutral and ionized atoms and molecular fragments from a solid surface. This fragmentation pattern can be interpreted much like a traditional MS spectrum, providing chemical specificity at a spatial resolution between 50 nm and 1 μm. 104 This technique is inherently surface-selective with a depth of penetration of ∼10 nm. Ion ablation, which can remove layers of material from a solid surface, effectively rendering a 3D chemical image, can extend the usefulness of SIMS. Time-of-flight secondary ion mass spectrometry (ToF-SIMS) can be utilized on biomass to produce spectra in which characteristic ToF-SIMS ion fragments of major cell wall components can be assigned, many of which are currently published. 133 –135 Jung et al. used ToF-SIMS to analyze the chemical composition of cross-sections of juvenile poplar (Populus deltoides) stems before and after dilute acid pretreatment. 136 It was determined that not only do the relative contents of cellulose, xylan, and lignin significantly change after pretreatment, but more importantly, the changes at the surface detected by ToF-SIMS do not match changes indicated by bulk compositional analysis. 136 ToF-SIMS showed that a much higher percentage of xylan remains on the surface of biomass after hydrothermal pretreatment, an observation considered important because cellulolytic enzyme activity is highly dependent on interactions with the substrate surface. 136 In a similar study investigating cellular spatial distributions with respect to lignin S/G ratio on Populus trichocarpa wood cross sections, this technique found that a higher proportion of G lignin resides in the middle lamella, while the inner cell wall area was much more S lignin rich. 137
Another useful MS application, MALDI-imaging MS (MALDI-IMS), can be used to determine the spatial distribution and relative abundance of polymers or macromolecules of a specific molecular weight or DP. 138 For example, the molecular weight spatial distribution of cellulose on cross-sections of juvenile poplar (Populus deltoides) stems after lignin removal by acidic sodium chlorite holocellulose pulping and hemicellulose removal by acid hydrolysis, was demonstrated by MALDI-IMS. 139 The results indicate a heterogeneous spatial distribution of cellulose molecular weight across the cell wall surface. 139 Unfortunately, MALDI MS techniques have been shown to discriminate against high molecular weight species in highly polydisperse samples due to differentials in ionization efficiency and detector saturation. 140 This may explain why the authors never obtained significant intensities for masses above ∼3,500 atomic mass units or cellulose DP of ∼20. Clearly, this limits the application of MALDI-IMS in studying naturally occurring plant cell wall polymer systems, which inherently have high polydispersities. However, the molecular images generated from MALDI-IMS can still provide spatial distribution information on low molecular weight oligosaccharides, a particularly useful data subset in biomass deconstruction studies. Lunsford et al. utilized a similar MALDI-IMS method on young Populus wood stems, demonstrating that tandem MS effectively reduces the background wood tissue ion signal, increasing S/N. 141
Coherent anti-Stokes Raman microscopy
There are several spectroscopy-based imaging techniques that have been applied to biomass to analyze the spatial distribution of various chemical features. These techniques are ideal for in situ conversion studies because they offer high chemical specificity in a non-destructive and real-time fashion without the need for labels. X-ray photoelectron spectroscopy (XPS) techniques have been widely used to analyze plant cell walls, although their use has been quite limited because of the lack of chemical specificity. 142 FT-IR microspectroscopy can provide the required chemical specificity, but is deficient in spatial resolution because of the long wavelength of infrared radiation and reduced penetration depth in high water content substrates like biomass. 143 Raman microspectroscopy, on the other hand, not only has excellent chemical specificity but also spatial resolution. 144 The Raman-scattering effect is particularly weak when compared to infrared absorption, requiring long pixel acquisition times, but recent studies have shown that effective real-time Raman-based imaging can be achieved via coherent anti-Stokes Raman scattering (CARS). 145, 146
Spectral contrast in CARS is still based on molecular vibrations—much like traditional Raman scattering—but CARS utilizes two laser light sources to generate anti-Stokes signals in a non-linear process. 146 The coherent Raman signal generated when the anti-Stokes signal is properly adjusted to match a particular Raman-active vibrational frequency is orders of magnitude greater than that from spontaneous Raman scattering. This produces the requisite signal intensity to image complex structures such as the plant cell wall and highly dynamic processes such as biomass conversion. Saar et al., using Raman bands representative of lignin and cellulose assigned in previous Raman spectroscopy studies, generated images of lignin and cellulose distributions in a variety of corn stover cell types and after delignification with stimulated Raman scattering (SRS), a technique closely related to CARS. 146 Raman vibrational modes at 1,600 cm−1, attributed to symmetric aromatic ring C=C stretching in lignin, and between 1,096–1,122 cm−1, attributed to the C–C and C–O stretching in cellulose, were used to image lignin and cellulose distributions in the biomass cross-sectioned samples. 146 In a subsequent study, Zeng et al. analyzed wild-type and down-regulated alfalfa lines and determined that the lignin content was much lower in the native sample, especially in the cell corners. 145
Mode-synthesizing atomic force microscopy
Atomic force microscopy (AFM), a type of scanning probe microscopy that gathers topological information pertaining to a substrate surface by a mechanical probe, has been employed to map nanoscale surface features such as biomass morphology and cellulose microfibril shape, dimension, and surface roughness. 104 An attractive feature of AFM is its high resolution; it is able to detect features on the order of a nanometer, several orders of magnitude better than optical techniques, due to the diffraction limit. Utilizing high-resolution AFM, Ding et al. proposed a new molecular model for the cellulose microfibril, suggesting that the elementary cellulose fibril has a core shell configuration composed of 36 glucan chains. 147 The most inner core has six true crystalline chains surrounded by 12 para- or sub-crystalline chains, followed by another shell composed of 18 more sub-crystalline or non-crystalline chains at the cellulose fibril surface. 147
Chemical characterization of plant cell walls on a nanometer scale is a much-needed methodology due to the heterogeneity and complexity of the plant cell wall and its small structural dimensions, but until recently has been a challenge. 148 Tetard et al. recently demonstrated the utility of mode-synthesizing atomic force microscopy (MSAFM) on Populus before and after a holopulping treatment and observed significant changes in the lamellar region of the holocellulose poplar cross sections. 149,150 MSAFM combines AFM with ultrasonic microscopy by monitoring the probe tip response with respect to high-frequency cantilever oscillation and sample oscillation due to mechanical excitation in the sample mount—generated in both cases by a piezoelectric crystal. 149,150 The signal extracted from the probe tip response is characteristic of the material composition, relaying nanoscale chemical and physical information.
IMAGING CELLULASE–CELLULOSE INTERACTIONS
Single molecule spectroscopy
Nature has devised a variety of strategies for the deconstruction of biomass; in the most common procedures to generate cellulosic ethanol, cellulases, secreted as “free” enzymes largely from fungi or bacteria, are utilized to depolymerize the cellulose locked into the lignocellulosic matrix of plant cell walls into fermentable monosaccharides. 4 Cellulases are complex enzymes composed of several components, including catalytic domain(s) and CBM(s), the synergistic activity of which enables effective hydrolysis. 10 The CBM is responsible for site-specific binding, disrupting target structures, aligning the polysaccharide chains, and enabling proximity to the catalytic domain(s). Enzymatic hydrolysis is the primary process bottleneck with respect to cost and yield in the production of cellulosic ethanol; as a result, having a deep understanding of the binding and hydrolysis activities of cellulolytic enzymes is important to making lignocellulosic biofuels cost-competitive. 10 Single molecule spectroscopy (SMS) approaches have therefore been applied to investigate the dynamics and kinetics of individual cellulases on cellulose surfaces. 151 SMS approaches for studying the binding and hydrolysis activities of cellulolytic enzymes include both optical and non-optical techniques, typically requiring high sensitivity and nanometer resolution.
Total internal reflection fluorescence (TIRF) microscopy uses the evanescent wave produced at the interface of liquid media and an attenuated total reflectance crystal to excite fluorescent tagged molecules in the narrow region in which the evanescent wave extends into the liquid (typically extending ∼100 nm). 151 The evanescent wave's intensity exponentially decays with distance from the surface of the crystal; as a result, the trajectory of the fluorescently tagged molecules can be tracked. Other SMS optical methods include photo-activatable fluorescent proteins (PA-FP) and defocused orientation and position imaging (DOPI), which uniquely allow for the determination of the 3D spatial orientation of an enzyme. 152,153 On the other hand, AFM offers a non-optical option that produces nanometer resolution topological descriptions of substrate surfaces via mechanical action (Fig. 6). 147,154
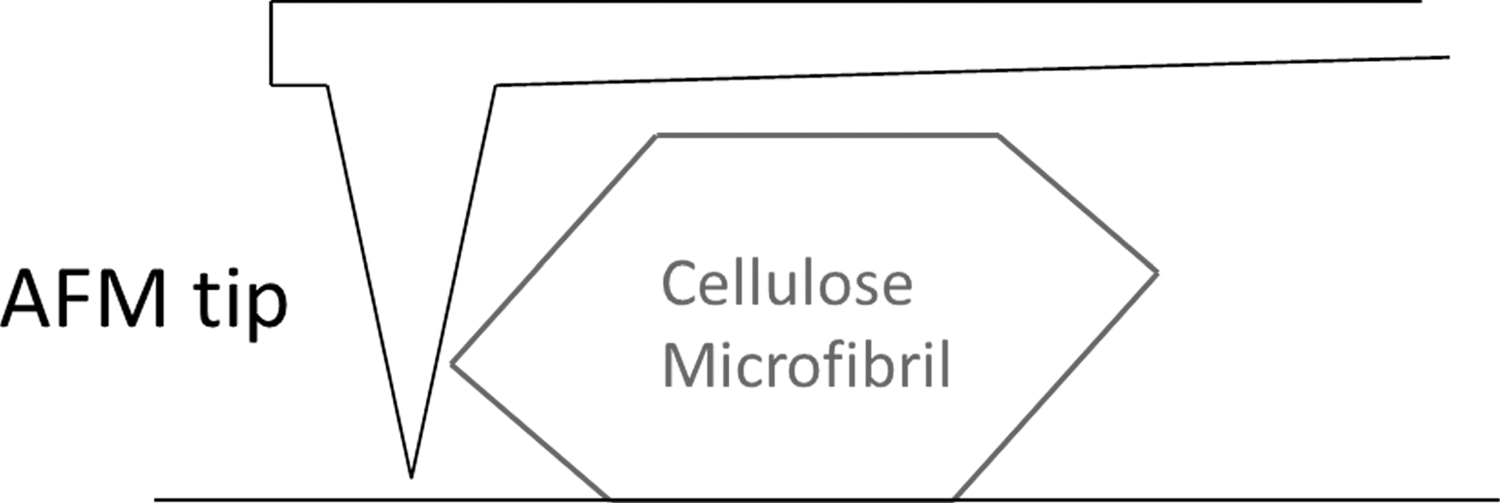
A schematic of AFM performed on a cellulose microfibril.
Liu et al. at NREL reported the first use of SMS techniques to investigate cellulose-degrading enzymes. 151 In this study, CBMs were bound to Valonia cellulose crystals followed by bio-conjugation with semiconductor quantum dots (QDs). By tracking individual QDs, it was determined that CBMs from Acidothermus cellulolyticus display linear motion along the long axis of the cellulose fiber despite increasing the CBM concentration from 25 nM to 25 μM. In a subsequent study by the same researcher, utilizing both TIRF and AFM, the analysis on green fluorescence protein (GFP)-labeled cellobiohydrolase-I (CBH-I) secreted by T. reesei confirmed the linear, directional motion on cellulose, while also indicating that cellulose crystallites change shape, decrease in width, and increase in surface roughness upon enzymatic attack. 153 In their most recent contribution, Liu et al. applied single molecule DOPI to extract 3D spatial binding information about GFP labeled family 1, 2, and 3 CBMs (fungal free cellulase, bacterial free cellulase, and bacterial cellulosome, respectively) bound to Valonia cellulose crystals. 152 In all cases, they observed that in situ CBMs attached consistently to the two “hydrophobic” planar faces of the cellulose crystal, displaying a well-defined angle between enzyme and crystallite. This indicates that CBMs have binding mechanisms driven by both chemical and structural recognition of the cellulose crystallite surfaces. 152
Fluorescence resonance energy transfer
Based on the known relationship between cellulase binding and cellulase activity, considerable efforts beyond SMS approaches have been made to examine and understand the fundamental interactions between cellulase and cellulose. Bulk, signal averaging techniques such as ultraviolet-visible spectroscopy, NMR, and quartz crystal microbalance (QCM) have been applied to probe cellulase catalytic activity, affinity, and binding properties on cellulosic substrates; however, few methodologies can offer the level of real-time, in situ information about cellulase–cellulose interaction as fluorescence resonance energy transfer (FRET). 155 –157 FRET has been applied to biological systems to study biomolecule interaction for some time, although Wang et al. demonstrated one of the first applications for investigating cellulase–cellulose interactions. 158 FRET utilizes labeled molecules containing a donor/acceptor fluorophore pair; interactions and trajectory of those labeled molecules are investigated by a FRET signal (Fig. 7). The FRET signal is resolved from the donor fluorescence emission, which is emitted upon donor excitation and only when the distance between the molecules labeled with the donor/acceptor fluorophores are within the prerequisite FRET distance (typically within ∼10 nm). This FRET signal is therefore also proportional to the distance between the labeled molecules.
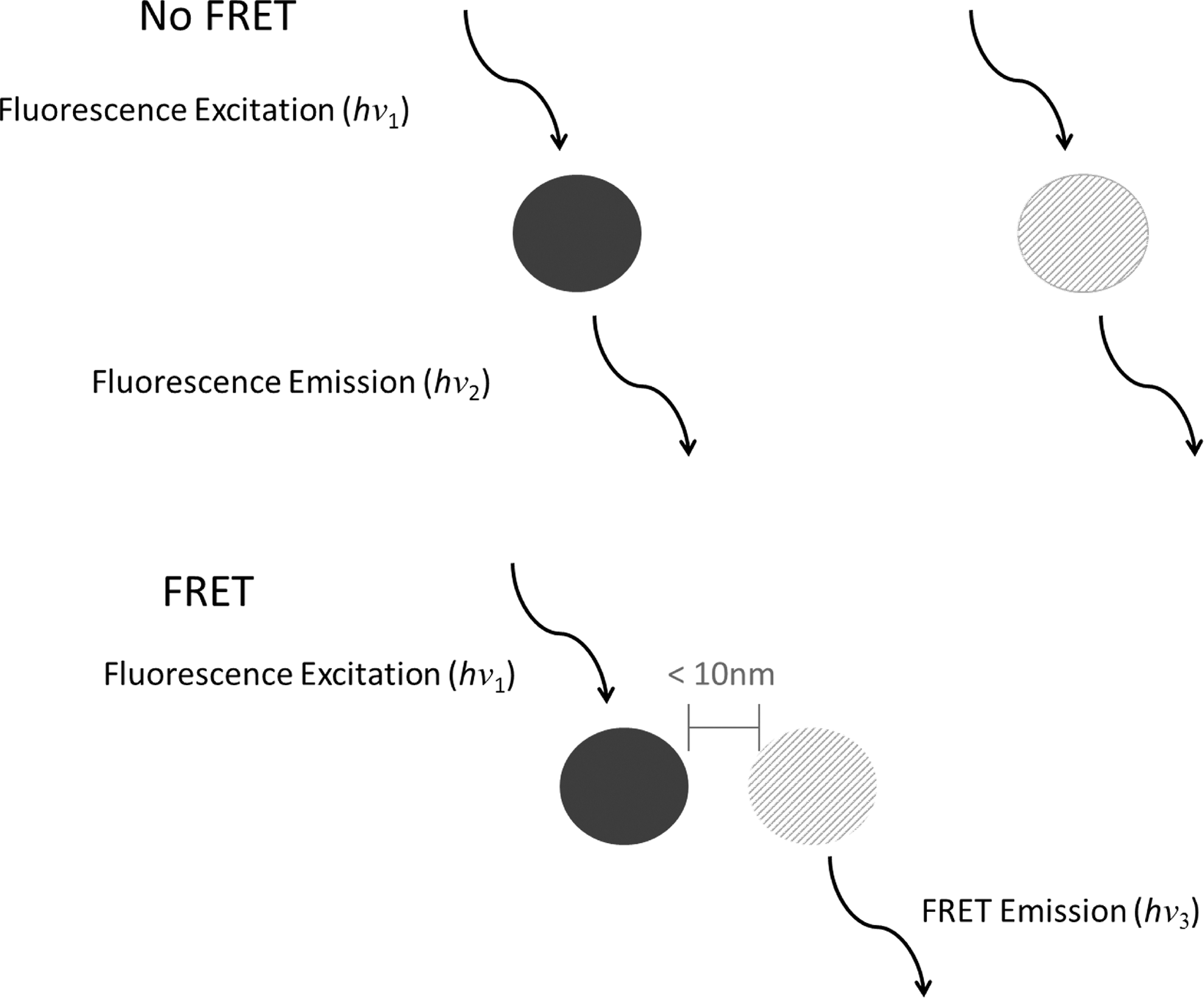
A schematic describing the distance dependency of FRET and the mechanism relating energy transfer between two chromophores.
Wang et al. reported covalent labeling of carboxymethyl cellulose (CMC) with a donor 5-(aminomethyl) fluorescein dye and cellulase from T. reesei with an acceptor AF594 dye, ultimately determining that lower temperatures seem to favor cellulase binding to CMC substrates. 158 In subsequent FRET microscopy studies from the same lab, labeled fluorophore pairs on cellulase—a mixture of endoglucanases, cellobiohydrolases, and β-glucosidases—from T. reesei found that cellulase binds in a non-uniformly distributed fashion along the length of a cellulose fiber with high concentrations at the ends of fibers. These findings supported similar work by Hilden et al. 159 This study seemed also to show that the proximate distance between FRET pairs is equal to the average length of the cellulase, suggesting that cellulase molecules can bind next to one another along the length of a cellulose fiber.
Conclusions
Lignocellulosic biomass is a multi-scale, complex, and highly heterogeneous material, the composition and structure of which vary widely depending on the source and on a range of genotypic, phenotypic, and environmental growth factors. Decades of research have been dedicated to understanding and deconvoluting the underlying phenotypic, biochemical, and molecular properties responsible for the ability of the plant cell wall to resist deconstruction. Though this research has been useful in efforts to commercialize the biological conversion of lignocellulosic biomass into materials and fuels, many conclusions have been based on observations of either isolated cell wall components or biomass with altered structure. Typically, this research also focuses on the effect of individual characteristics and fails to take into account the combined and integrated effect of an array of cell wall characteristics on recalcitrance; in some cases, this has led to conflicting or even erroneous conclusions. Though some characteristics are most likely more influential than others, the relationship between substrate characteristics and recalcitrance is clearly multi-variant and non-linear, thus virtually impossible to appreciate fully in the absence of a comprehensive characterization.
Moving forward, a useful strategy to identify the mechanisms of and overcome biomass recalcitrance will include the generation of better cell wall models through continued investigations of cell wall biophysics and biosynthesis; the identification of samples from a wide range of sources with extreme sugar release phenotypes in combination with extensive genomic and proteomic characterization; and, most importantly, a multiple length scale characterization of reduced-recalcitrance substrates, transgenics, etc., for analyzing defined collections of potential recalcitrant-related chemical and molecular features. This, however, is unlikely to occur in any single study generated by an individual investigator. It will require assembling specific expertise from multiple institutions and providing that group with the means to collaborate effectively across time, distance, disciplines, and interests. The US Department of Energy Bioenergy Research Centers, composed of multidisciplinary teams of scientists, is an ideal model for this challenge.
In addition, improved methodologies to characterize the chemical and molecular structures related to biomass recalcitrance, i.e., cell wall component 3D ultrastructure and organization and the interactions between biomass and enzymes, are needed. Techniques to probe the effects of substrate surface properties and enzyme structure on the properties of enzyme–substrate interactions in the biological conversion of cellulosic biomass are particularly under-developed and under-utilized. Though these improved methods can facilitate considerable progress, major advances in understanding the fundamental features of lignocellulosic biomass could simply be made if more comprehensive research studies were conducted.
Footnotes
Acknowledgments
This work was supported and performed as part of the BioEnergy Science Center, managed by Oak Ridge National Laboratory (ORNL, the manager partner and home facility for the BioEnergy Science Center). The BioEnergy Science Center is a US Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. ORNL is managed by UT-Battelle, LLC, under contract DE-AC05-00OR 22725 for the US Department of Energy.
Author Disclosure Statement
No competing financial interests exist.