
Abstract
Food can be used as a delivery vehicle for nutrients and other active compounds to promote health and prevent diseases. Encapsulation in microparticles provides the opportunity to enhance the stability of nutrients during processing and storage and could potentially improve their uptake by tissues in the gastrointestinal tract. This work describes the use of a simple soft lithography-based protocol for the fabrication of monodispersed polymeric microparticles with precise, non-spherical shapes. A variety of microparticles with different morphologies were fabricated using poly(lactic, glycolic) acid, polycaprolactone, the pH-sensitive polymer Eudragit®-S100, and pectin-alginate mixtures. Vitamin C and anthocyanins were used as model nutrients for encapsulation to enhance food nutritional value and appearance. The results illustrate the applicability of this high-throughput microfabrication method to obtain microparticles with precise control over size and shape using low processing temperatures to avoid damage of nutrients.
Introduction
Biodegradable polymeric microparticles have been successfully studied as delivery vehicles for pharmaceutical compounds and other active components for medical treatments. 2 Microencapsulation allows more efficient release profiles with localized and controlled delivery over time. Methods for obtaining spherical microparticles include spray drying, emulsion-solvent evaporation, air atomization, and liposomal encapsulation, among others. 3 –9
Although micro/nanoparticles are the gold standard for drug delivery, their application in the food industry has not been fully explored. Microencapsulation of bioactive compounds such as vitamins and other food additives is a promising approach to offer protection from the acidic environment of the stomach, avoid oxidation, and enhance stability within the intestine for prolonged and controlled release. Additionally, when used for food additives, microencapsulation has the potential to avoid deleterious reactions in food mixtures, thereby enhancing their stability during processing and storage and avoiding undesired changes in color or taste. 1
Recent advances in micro/nanotechnology have allowed the fabrication of drug delivery devices with highly uniform sizes and well-defined symmetrical and asymmetrical structures that are impossible to obtain using conventional microparticle manufacturing methods, providing the devices many attractive features for oral delivery. 10 –12 Nevertheless, these fabrication processes present several disadvantages: cleanroom-based techniques are expensive; there is a limited pool of materials to work with; and they may entail damaging processes (corrosive etches, toxic solvents, and elevated temperatures). Most importantly, the high cost associated with the materials, facilities, and processing would be a limiting factor for the practical implementation of this technology, especially in the food industry. As a rough estimate, the fabrication of microparticles via cleanroom-based methods would increase the cost of the encapsulated nutrient by hundreds of dollars per milliliter, even at mass production scale. Microparticle fabrication techniques for producing microspheres do not provide precise control over particle geometry and dimensions. These methods are restricted to the fabrication of spherical or irregularly-shaped particles, and in most cases, produce batches with wide size distributions. 3,6,9
Many lower cost techniques have been developed or are under development for polymer microfabrication. These typically possess greater versatility in materials and processing approaches than silicon-based microfabrication techniques. Soft lithography is a group of techniques that uses an elastomeric stamp with topological microfeatures to generate micro- or even nanostructures. 13 Soft lithography provides simple, versatile, and cost-effective fabrication methods for creating microparticles with precise control over size and geometry. 13 The use of soft lithography can reduce the cost of encapsulation of precisely defined particles down to less than one dollar per milliliter for mass-produced particles.
Using these fabrication techniques microparticles can be made from polymeric materials with different degradation rates, and their shape and size can be tailored to increase interaction with intestinal tissues by changing their dynamic properties. 14 The nutrients can be delivered as the polymer starts to degrade in response to environmental changes (e.g., pH, temperature, magnetic field, ion concentration). 12 Additionally, uniform particle size distribution and morphology allow better control over the release kinetics of encapsulated molecules.
Soft lithography has been used to develop versatile and simple procedures to fabricate polymeric microparticles. 15,16 These fabrication processes allow for the production of monolithic microparticles with a uniform blend of solid polymer and solid active compound, microparticles with reservoirs, and self-folding microparticles for increased attachment to soft tissues. The versatility of these fabrication methods has been demonstrated using a host of materials of interest to the medical and food industries including: hydrophobic polymers such as polycaprolactone (PCL); hydrophilic polymers such as poly(lactic, glycolic) acid (PLGA); hydrogels such as co-polyethylene (glycol) diacrylate (co-PEGMA) and poly(ethylene glycol) dimethacrylate (PEGDMA); and cross-linked polysaccharides such as chitosan. This array of materials allows for the incorporation of varying amounts of a range of hydrophobic, amphiphilic, and hydrophilic molecules into the microparticles.
Briefly, PLGA, chitosan, and poly(PEGMA-co-PEGDMA) monolithic microparticles are molded using previously patterned poly(dimethyl siloxane) (PDMS) molds with different microfeature arrays. After solvent evaporation, and due to the presence of discontinuous dewetting, the polymer is deposited on selective portions of the mold. 17 Finally, the dried polymer is removed from the PDMS mold by stamping it on top of a poly(vinyl alcohol) (PVA) sacrificial layer at temperatures and pressures in the range of 80–120°C and 30–90 KPa, respectively. The polymer is subsequently dissolved in water to release the particles. 15 Following a similar fabrication protocol, self-folding microparticles with single and multiple reservoirs can be obtained as bilayered structures from PLGA, chitosan/poly(PEGMA-co-PEGDMA), or poly(methacrylic acid)/PEGDMA. The combination of polymers with different swelling profiles allows volume expansion-induced self-folding. The applicability of these self-folding microstructures for transmucosal drug delivery has been initially assessed using a pig intestinal mucosa model. 16
Here we describe a fabrication protocol that can be applied to a variety of natural and synthetic polymers (e.g., pH-sensitive polymers, hydrogels, hydrophobic and hydrophilic polymers, and polysaccharides such as pectin and alginate). In contrast to previous reports on soft lithography-based production of microparticles, the methods described here not only allow for the fabrication of microparticles with precise control over size and geometry in a high-throughput manner, but also use significantly lower processing temperatures and pressures, which reduces the overall manufacturing cost and provides more benign conditions for the encapsulated bioactive agents. The application of these methods for nutrient encapsulation for potential delivery was assessed using Vitamin C and anthocyanins (ACN) as model active compounds.
Materials and Methods
Materials
A novel fabrication protocol has been developed to produce pH-sensitive microparticles from Eudragit S100 (ES100) (Evonik, Essen, Germany). ES100 consists of anionic copolymers based on methacrylic acid and methyl methacrylate and has been previously used for the fabrication of carriers of a number of bioactive compounds such as Vitamin C and piroxicam. 4,5,18 For our studies, Vitamin C, a well-known antioxidant and food additive, was used as a model bioactive compound. ES100 is a biocompatible copolymer that maintains a compact structure in acid environments but becomes soluble at basic pH values. ES100 is thus an excellent material for the encapsulation of active components that need to be protected as they pass through the gastrointestinal tract and are released at the small intestine for absorption. The application of this fabrication protocol was further extended to biopolymers approved for food applications (e.g., pectin-alginate), and pH-sensitive formulations were developed.
ES100 polymer was provided by Evonik, alginate (Protanol SF120RB) was provided by FMC Biopolymer (Philadelphia, PA), and high methoxy rapid- and slow-set pectins (Pretested) were provided by TIC Gums (White Marsh, MD). Red cabbage extract was purchased from RFI Ingredients (Blauvelt, NY) and the glucono-δ-lactone (GDL) was purchased from Tokyo Chemical Industry Co, Ltd (Tokyo, Japan).
Microparticle Fabrication
A 3–5% ES100 solution in isopropanol-water (9:1 ratio) containing 33–51% vitamin C (w/w) was spin coated at 4,000 rpm for 60 sec on top of a previously patterned PDMS mold with different types of microfeatures (e.g., circular or hexagonal microwells). The excess polymer on top of the PDMS mold was manually stamped out on top of a PVA-coated substrate (e.g., glass or silicon wafer) at 40–45°C (first stamp). The remaining material inside the PDMS microwells was subsequently transferred onto another PVA-coated substrate using a hot embossing system (EVG®520IS, EVG, St Florian am Inn, Austria) at 0.75–1.5 MPa and 33–40°C for 5–10 minutes (second stamp). Prior to each stamping step, the PVA-coated substrates were exposed to steam to increase the adhesion between the polymer and the PVA layer. As previously described, the PVA coating was used as a sacrificial carrier layer for later release of the microparticles in an acidic aqueous solution. Alternatively, the microparticles were released from the PDMS mold under more benign conditions (e.g., at room temperature) using a sacrificial layer lift-out approach in which a polymer solution–containing a solvent that does not dissolve the microparticle material–was cast over the PDMS mold, allowed to dry, and then manually peeled off along with the microparticles (Fig. 1). The loaded microparticles were subsequently released in a solution that specifically targets/dissolves the sacrificial layer material, such as an acidic aqueous solution for the ES100 microparticles lifted out with a PVA sacrificial layer.
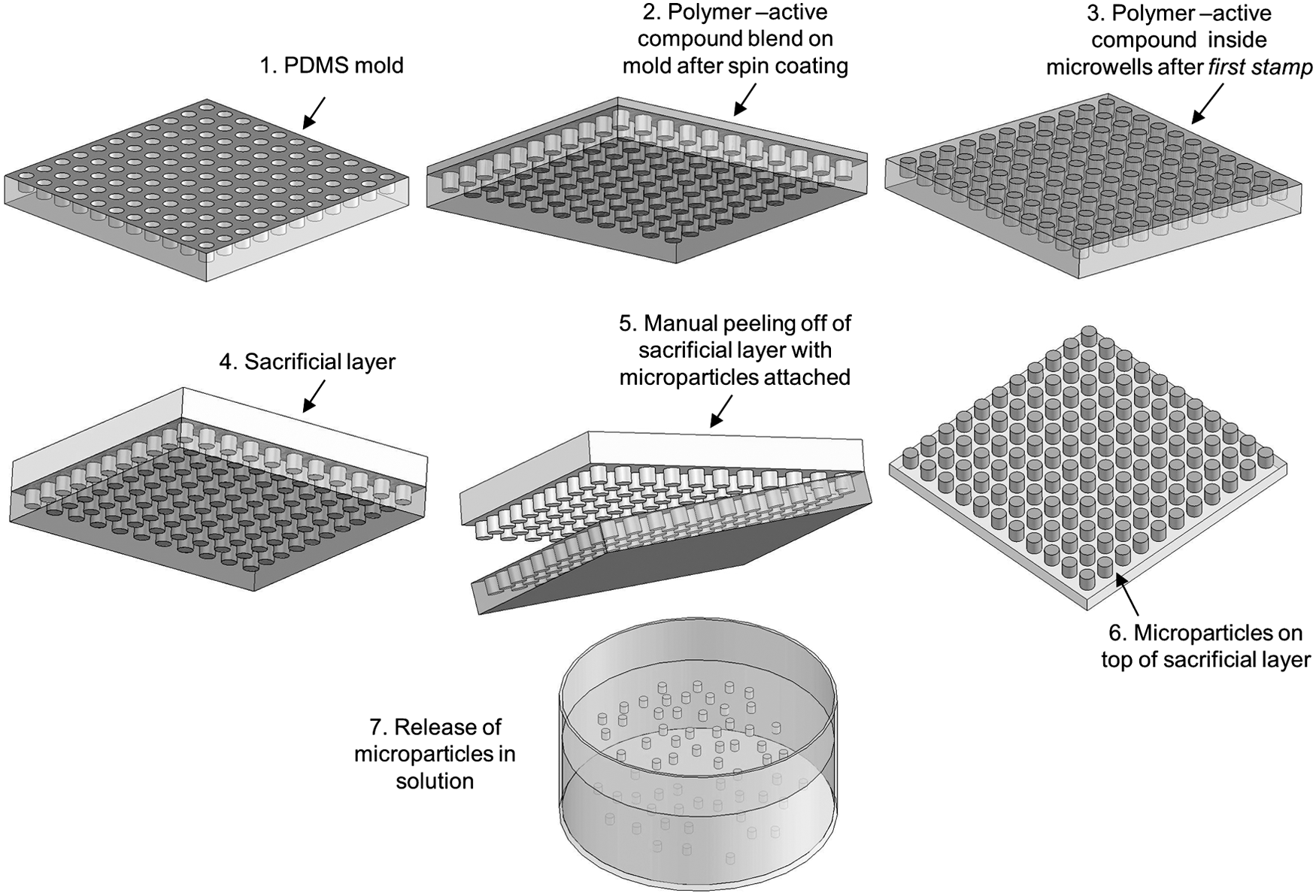
Schematic diagram of the lift-out molding fabrication protocol.
For the alginate-pectin microparticles, the PDMS molds were initially treated using oxygen plasma to increase the wettability of their surface for increased filling of the microfeatures. Subsequently, an aqueous alginate-pectin solution (0.769% alginate, 0.769% pectin, and 10% ethyl alcohol) was prepared. ACN solution was added to the mixture to a final concentration of 7.7% (v/w), and the mixture was stirred for one minute. Finally, GDL solution was added to the mixture to a final concentration of 2% (w/v), and the mixture was stirred for one additional minute. This hydrogel mixture was cast on top of the previously patterned PDMS mold. The PDMS mold with the alginate-pectin solution was placed in a vacuum desiccator for 400 min to improve loading inside the mold microwells. The remaining alginate-pectin solution outside the microwells was removed from the mold via spin coating (2,000 rpm for 45 sec). The polymer inside the microwells (alginate-pectin microparticles) was removed from the mold using a polystyrene (PS) sacrificial layer. This PS layer was deposited by spin coating a 25% PS solution in anisole at 600 rpm for 45 sec. The solution was dried at room temperature and then peeled off the PDMS mold. The alginate-pectin particles were attached to the sacrificial PS layer, which was dissolved in anisole to release the microparticles. The microparticles were then collected and resuspended in acidified water (pH 1.5).
Rheological Studies of Alginate-Pectin Mixtures
Rheological studies were performed to determine the alginate-pectin mixture gelling time and the strength of the formed hydrogels. Dynamic rheological analysis was performed on samples using a controlled stress rheometer (AR 1000-N, TA Instruments, New Castle, DE). A 60-mm acrylic cone was used for all experiments. A 4 mL sample of the gel mixture was placed on the rheometer plate. Mineral oil was used to coat the edge of the sample to prevent evaporation of water during the gelation process. Throughout the experiment, temperature was kept constant at 23°C and the stress and frequency were maintained at 0.1768 Pa and 1 Hz, respectively. Time sweeps were conducted every two minutes over a 4-hour period. The storage modulus, G′, and loss modulus, G″, were measured at each time interval. The sol-gel time was determined by taking the crossover point of the G′ and G″ curves. The final gel time was determined to be the point when the G′ and G″ curves reached an asymptotic value. The gel strength was determined as the G′ value that corresponded to the final gelation time.
Microparticle Characterization
Particle size and morphology were characterized via optical microscopy and scanning electron microscopy (SEM). All the steps of the fabrication process were documented via SEM, including images captured of the polymer removed after the first stamp, the particles inside the PDMS molds, the particles attached to the polymeric sacrificial layer, and the particles after being released from the sacrificial layer. Before SEM, all the samples were coated with a thin layer (∼10 nm) of gold-palladium to make their surface conductive. Particle size (i.e., diameter and thickness) was measured from a number of SEM micrographs (from at least two independent samples) using ImageJ software (National Institutes of Health, Bethesda, MD) and compared to the microscale geometry of the PDMS molds. Particle size distribution was also characterized by dynamic light scattering (DLS) (BI-200SM, Brookhaven Instruments, Holtsville, NY) using an elapsed time of 4:01 minutes, a first and last delay of 200.00 and 10.0 μsec, respectively, and 4.82 e+06 samples. The particle yield was calculated using ImageJ as the percentage of particles recovered from the PDMS mold on the sacrificial polymeric layer for 17.35cm2 PDMS molds containing approximately 1.97e+06 particles with 15-μm diameter.
Results and Discussion
Overall, the soft lithography fabrication method described in this work provided an average particle yield of 90.8±1.9% for microparticles prepared using ES100 and pectin-alginate blends. The high yield provides convincing evidence for the potential of the process for scale-up. The fabricated microparticles exhibited a uniform morphology and a narrow size distribution as confirmed by SEM (Figs. 2 and 3) and the size distribution curve obtained by DLS (Fig. 4). It is important to note that the average particle diameter obtained via DLS was smaller than that measured via SEM due to the cylindrical rather than spherical geometry of the particles analyzed.

SEM image of excess polymer (ES100) removed from the PDMS mold (with 10-μm diameter microwells) after the first stamp

Micrographs of microparticles fabricated using the lift-out method. Alginate-pectin gel microparticles on a PS lift-out layer (15-μm diameter)
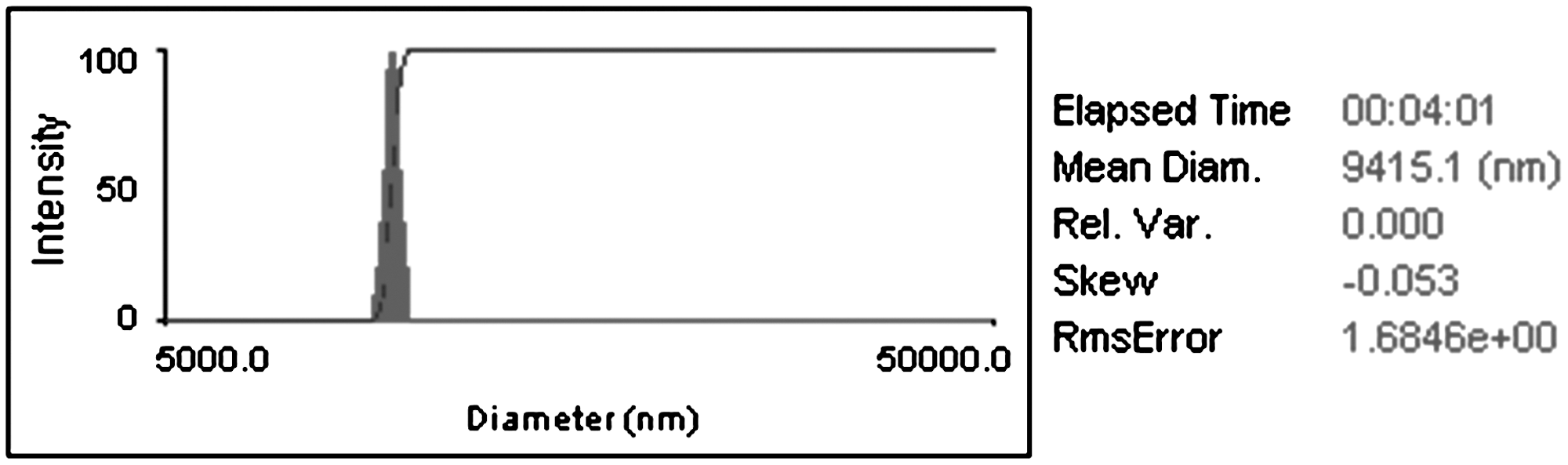
DLS results of particle size distribution of cylindrical microparticles (15-μm diameter and 3-μm height, containing ∼51.6% of vitamin C) suspended in acid solution (pH∼1).
The microstructure on the PDMS mold was accurately reproduced for all the polymers used in this study. Microparticles fabricated using a mold with 15-μm diameter microwells had an average particle diameter ranging from 13.34±0.08 to 15.83±0.10 μm for pectin-alginate and ES100 microparticles, respectively (Fig. 3F). The microparticle thickness can also be controlled by changing the polymer solution and the spin-coating parameters (Table 1).
Thickness Measurements for Particles Fabricated Using Different Polymer Solutions and Spin-Coating Parameters on a PDMS Mold Containing an Array of 15-μm Diameter Wells
One of the major limitations when implementing standard cleanroom- or non-cleanroom-based microfabrication techniques in the food industry is the lack of established fabrication protocols for materials that are approved for food applications. Natural materials such as pectin and alginate have been widely used in the food industry due to their physicochemical properties and positive physiological effects. 19 As such, we adapted our fabrication protocol to be able to produce pectin/alginate microparticles loaded with ACN. ACN are plant pigments with strong antioxidant properties that can provide appealing colors to foods and beverages while enhancing health, particularly of the GIT. 20
The gel formation of pectin-alginate mixtures is controlled by the pH of the solution. For alginate-pectin mixtures, it has been reported that gelation does not occur until the pH of the mixture drops below 3.4. 21 Toft et al. reported that mixtures of alginate and high methoxy pectin formed thermo-reversible gels below pH 3.8. 22 These studies used GDL as a slow acidifier to regulate the pH decrease.
In the study reported here, we added GDL at 2% (w/w) to control the pH decrease and gel formation concomitantly. Because GDL changes the pH of the mixture as a function of time, gel formation was monitored by recording the rheological properties of the mixture as a function of time. The rheological properties of the polymer blend will influence the available processing time to manufacture the microparticles, and the gel strength may influence the microparticle release profile. Therefore, we characterized the rheological properties of the gel blends used, observing the viscous (G″) and elastic (G′) moduli as a function of time post-mixing (Fig. 5). The pH of the solution plays an important role both in gel formation and in ensuring that the ACN remains unoxidized during processing, so it must be monitored as a function of processing time. Fig. 5A shows the superimposed rheological and pH data for a 50-50 blend of alginate-low methoxy pectin, showing that the solution remains workable for more than 100 minutes (at the crossover point of G′ and G″) and the corresponding pH at the start of gel formation and at the end of gel formation, where the G′ reaches to an asymptote. By monitoring pH, it is also apparent that pH remains low enough throughout the entire process to maintain the ACN in its unoxidized state.
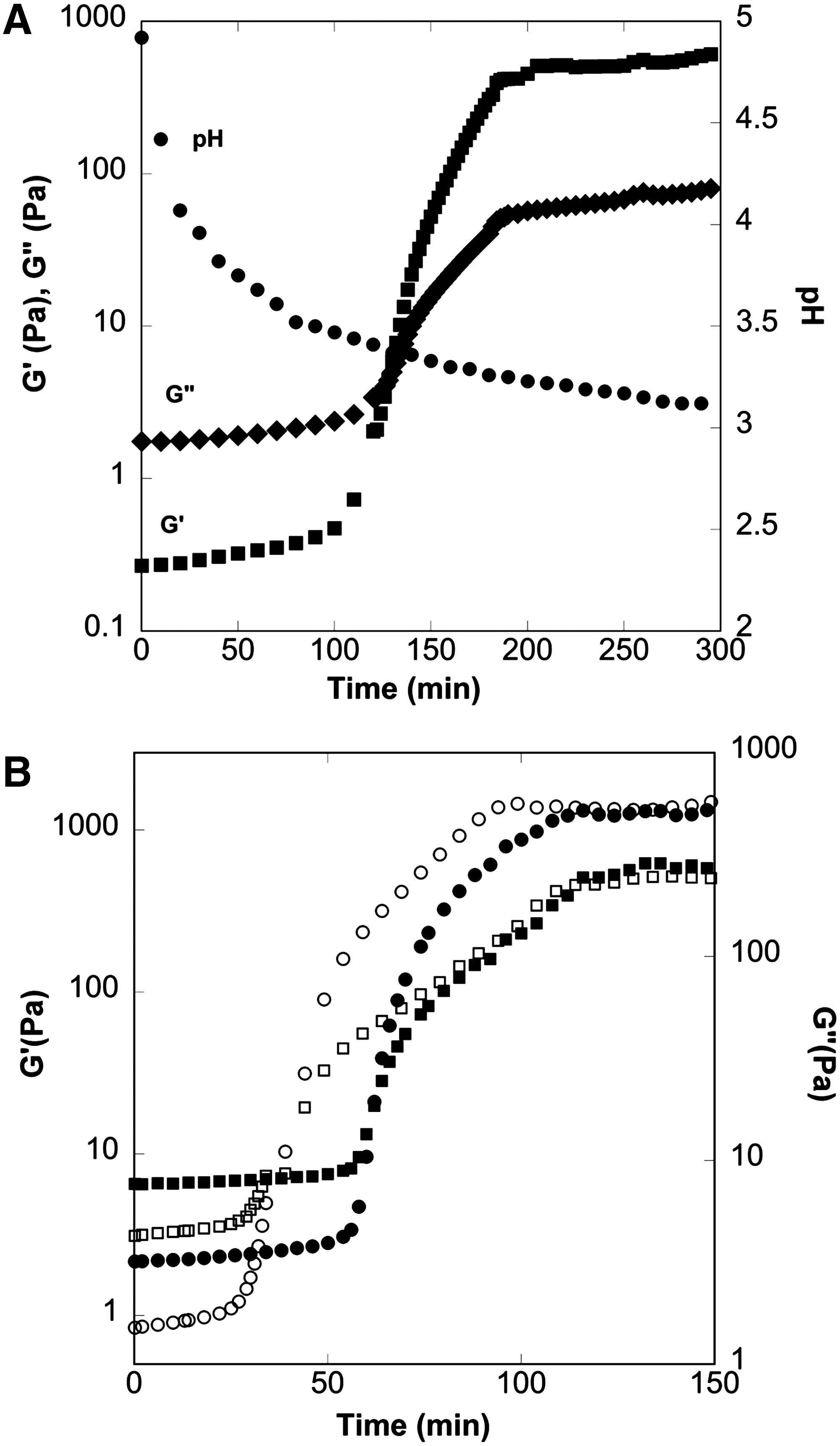
The pH values of the initial alginate and pectin solutions were measured to be 5.78 and 3.17, respectively. Therefore, depending on the ratio of alginate-to-pectin and the type of pectin used (e.g., low methoxy, rapid- or slow-set high methoxy), the initial pH values of the mixtures were different. The pH of the initial mixture affected the final pH of the gel and the gel formation time (Table 2). Fig. 5B shows that, for a mixture of sodium alginate and high methoxy pectin, changing the ratio of alginate to pectin from 50:50 to 80:20 increases the gel starting time from approximately 40 to 60 minutes for the same GDL concentration. However, the gel starting pH is independent of the alginate-to-pectin ratio, with an average value of 3.57±0.1 (Table 2). The gel start pH was determined to be between the pH values reported by Morris and Chilvers and by Toft et al. 21,22 When the GDL concentration was increased, the gel starting time and final gel pH were reduced (data not shown). This information shows that processing conditions should be adjusted to compensate the formulation changes and can be utilized for optimization of processing conditions and encapsulant formulation.
Rheological Evaluation of Gels Produced Using Different Alginate-Pectin Concentrations and Types of Pectins
Conclusions
In this paper we demonstrated simple, versatile, and comparatively inexpensive fabrication protocols for nutrient encapsulation in pH-responsive polymers and pH-responsive biopolymer blends approved for food use. The precise control over size, geometry, and bioactive compound quantities at the single particle level may provide more controlled and predictable release profiles. Furthermore, having precise control over microparticle shape and dimensions is one of the major advantages over conventional spherical fabrication methods, because these features can be adjusted to increase the active compound uptake into specific target tissues and to improve nutrient delivery procedures. Utilization of high-throughput methods together with food-approved biopolymers as encapsulants will present opportunities for novel applications of precisely shaped and sized microparticles in the food industry.
Footnotes
Acknowledgments
The authors would like to thank Evonik, FMC Biopolymer, and TIC Gums for providing polymers for this study as well as the Ohio Nano Tech West Laboratory staff for technical assistance. Funding was provided by a Food Innovation Center (The Ohio State University, Columbus, OH) grant and United States Department of Agriculture NIFA grant #2012-67017-30169.
Author Disclosure Statement
No competing financial interests exist.