
Abstract
Many algae species exhibit phenotypes that are of great interest for industrial applications. Systems biology approaches including large-scale metabolic reconstructions can guide and enhance production of biobased commodities such as fuel and human therapeutic proteins. Here we review the existing computational models of metabolism in algal species, as well as the databases and tools that are available to improve the sustainability of algae-to-energy production at industrial scales. As this field has progressed with genome sequencing, computational techniques, novel annotation tools and databases, systems biology will continue to evolve to advance the appeal of algae to a wide array of industries.
Introduction
Microalgae fossil records extend back more than three billion years, and over that period algae have become a vital and ubiquitous component of the Earth's biosphere. 1 Their importance is primarily due to their high photosynthetic productivity, which creates the foundation for many ecosystems. 2 With more than 40,000 algal species currently identified, microalgae have evolved to succeed in most environments on earth. 3 Many microalgal species are autotrophic, meaning they convert sunlight and CO2 into biomass, and include types of green algae (Chlorophyta), yellow-green algae (Xanthophyta), golden algae (Chrysophyta), red algae (Rhodophyta), diatoms, and dinoflagellates. 4 This diversity of species contributes to much of the current interest in using microalgae for a variety of commercial purposes, ranging from nutraceutical applications to biofuel production. Here we focus our discussion on eukaryotic species of algae and exclude cyanobacteria, or blue-green prokaryotic algae, due to their distinct cellular properties.
Industrial production strategies have generally sought to capitalize or improve production of one or more of the following outputs from algae: long-chained polyunsaturated fatty acids, food colorants, animal feed and supplementation, cosmetics, hydrogen, wastewater treatment, CO2 fixation, human therapeutic proteins, and biofuel production. 5 –13 Thus, algal species have the potential to become “cellular factories” upon genetic re-engineering to improve production and yield of desired commodities.
Despite the promise of commercial applications for microalgae, algal biotechnology remains in its infancy. Terrestrial crops and animals have been bred over thousands of years and selected for the creation of high-yield species for agricultural needs. It stands to reason that efforts to cultivate algae on a large scale will not be based solely on wild-type species but will require some degree of genetic modification before algae can make economically viable contributions to any sector. 14 However, our current limited understanding of how to perform genetic engineering of a species for the creation of target strains with economically favorable characteristics has hindered the industrialization of algal biotechnology.
Academic and commercial interest in the properties of algae metabolism has increased substantially in recent years (Figs. 1A-B). Advances in genome sequencing have allowed for the sequencing of at least 20 microalgal genomes, including the smallest free-living eukaryote Ostreococcus tauri, the unicellular green algae Chlamydomonas reinhardtii, and the planktonic species Botryococcus braunii, with applications across a range of industries (Fig. 1C). 9,15 –76 Key species of interest include B. braunii, C. reinhardtii, Dunaliella salina, and Phaedactylum tricornutum, although to date only a select number of engineered strains and products have been commercialized.
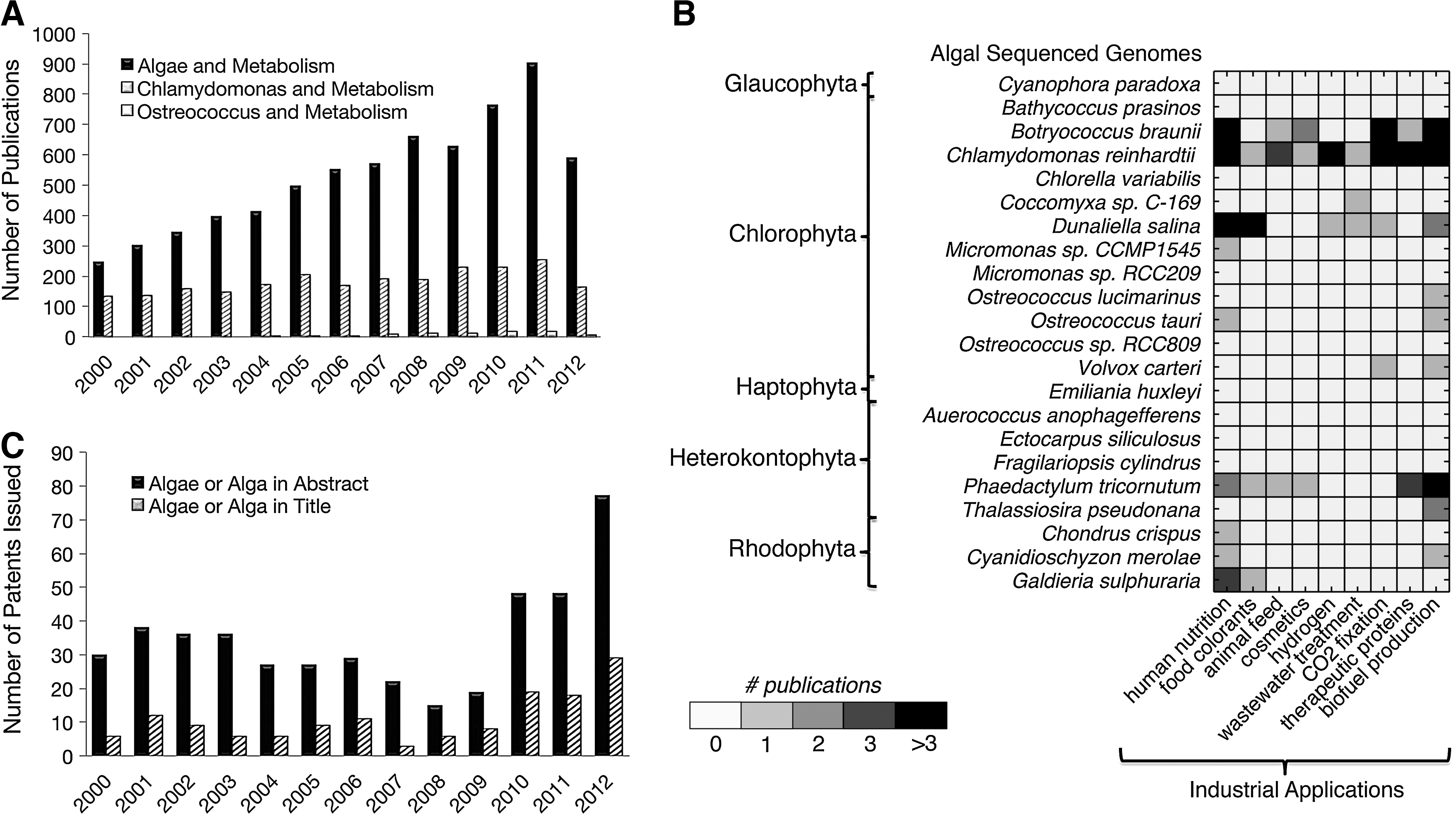
Snapshot of algae species with sequenced genomes in research and industrial applications. Number of publications cited in PubMed per year from 2000 through 2012
Furthermore, within the broader field of systems biology, advances in whole genome sequencing have paved the way for the reconstruction of organism-specific metabolic networks, with genome-scale networks having been created for several prokaryotes as well as for an increasing number of more complex, eukaryotic species. 77 Metabolic network models are systems-level, genomic computational tools that can assist in the reengineering of key metabolic pathways for industrial-level production of desired products such as lipids and biofuels. 78 Additional applications include bioremediation, wastewater treatment, and carbon dioxide (CO2) fixation. For example, a genome-scale metabolic network for the model eukaryote Saccharomyces cerevisiae was probed using in silico engineering strategies to increase ethanol yields on glucose and production of fumaric acid, a potential petroleum substitute. 79,80 Similarly, metabolic network reconstructions of algae hold promise to aid in the identification of genetic modifications for the creation of optimized strains with commercial potential for desired commodity production. To date, metabolic network reconstructions have been generated for four different algal species: C. reinhardtii, Ostreococcus lucimarinus, O. tauri, and B. braunii.
In this review, we highlight the current state of algae metabolic network modeling and optimization tools with an emphasis on the application of in silico metabolic engineering reconstructions. We use this information to provide an analysis of how systems-based approaches can inform and be informed by large-scale supply chain constraints in the algae-to-energy landscape, and how this might influence commercial development strategies.
Metabolic Engineering Reconstruction Process
Metabolic network reconstructions are mathematically organized structures assembled from various data sources including biochemical, genomic, and cell phenotypic data. Assembled networks account for known reactions, enzymes, and genes that are part of the metabolic pathways of an organism. Generating a metabolic network reconstruction is an iterative process involving several rounds of manual curation and experimental validation. The first step in creating a genome-scale metabolic reconstruction is to obtain the most recent version available of the target organism's sequenced genome and annotation to identify open reading frames (ORFs) and assign them functions by comparing them to genes associated with proteins of known functions using available online tools such as Uniprot and ChlamyCyc. 81,82
Once these molecular functions have been assigned, reactions are then associated with the annotated genes, taking into account stoichiometry and reversibility, using databases like KEGG and PredAlgo. 83 –85 Additionally, to create the most biologically accurate model with accurate network connectivity, cellular localization must be taken into account, especially for complex eukaryotic organisms such as algae. Unlike the other steps of the reconstruction process, which rely heavily on the comparison of the functionality of genes across different species and organisms, compartmentation of reactions requires organism-specific knowledge; however, there is often little to no evidence for reaction localization. 86 Thus, in the event that the localization of a particular reaction is unknown, researchers typically infer the compartment based on related reactions in homologous species, relying on transporter reactions to carry metabolites from one compartment to another. In the case of the well-studied plant species Arabidopsis thaliana and its reconstructed network AraGEM, compartmentation of reactions was completed manually based on the literature, and in many cases, reactions were localized by default to the cytosol to avoid relying on uncharacterized transport reactions. 86,87 Certainly, as biological knowledge increases, so too will the quality of available network reconstructions and associated reaction localization.
There are several freely available tools that semi-automate the draft reconstruction process, such as Pathway Tools and Model SEED; however, extensive manual curation is still required. 88 –90 Table 1 highlights key resources publicly available for the reconstruction of metabolic networks, including several algae-specific annotation tools. 81,82,85,88 –104 Ultimately, this extensive and iterative manual curation process results in an organism-specific, genome-scale metabolic reconstruction that summarizes the metabolic pathways of a target organism by documenting information such as the gene-protein-reaction (GPR) relationships, reaction stoichiometry, reaction reversibility, and cellular compartment. We provide an overview of the process to generate an in silico metabolic network for algae commodity production, including computational tools for optimized strain selection, in Fig. 2.
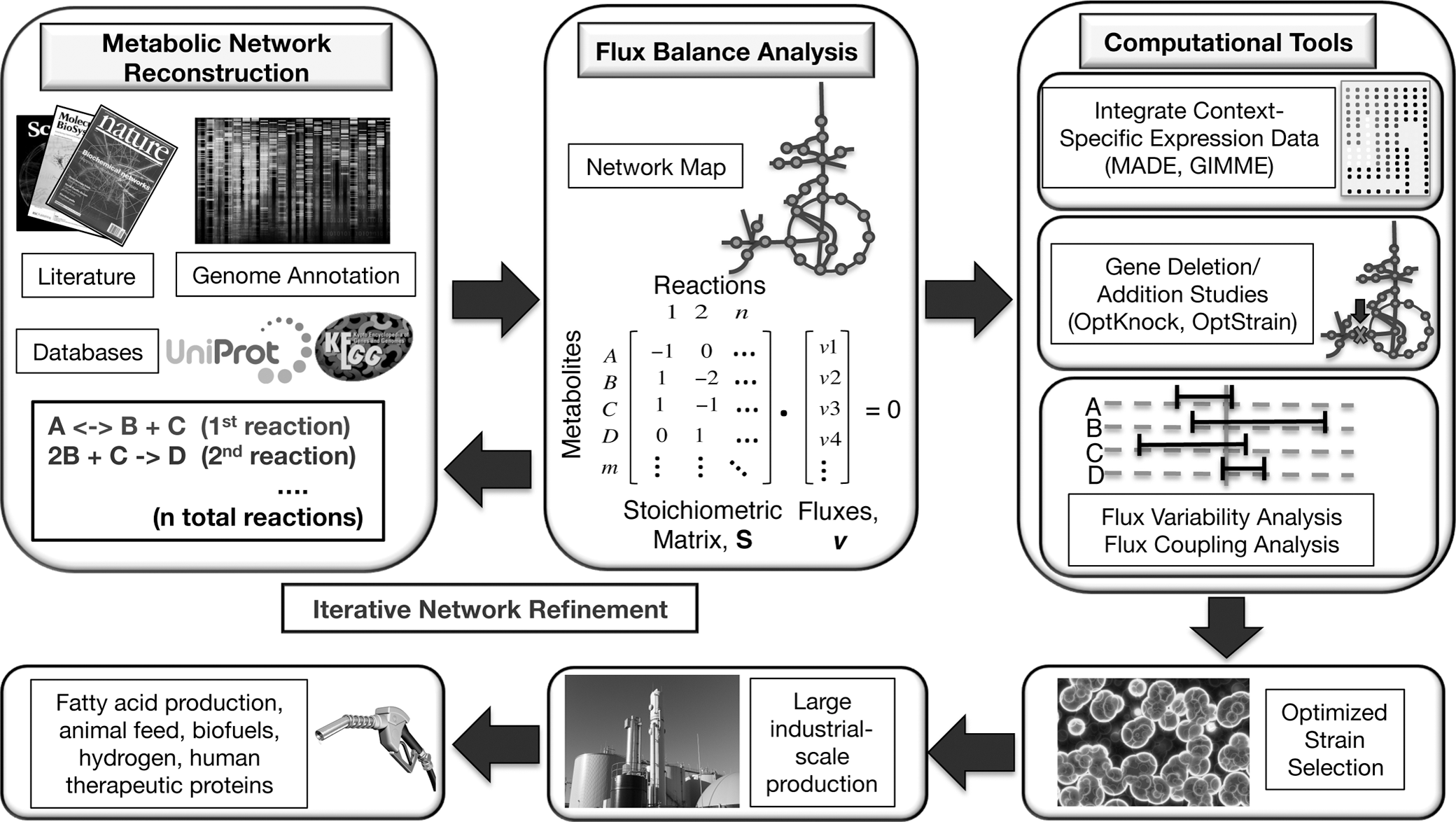
Snapshot of the in silico metabolic engineering process of algal strains for industrial commodity production. Genome sequencing allows for network formation and mathematical representation by a stoichiometric matrix. Computational tools may aid in the selection of engineered strains for large industrial-scale commodity production.
Genome and Pathway Resources Available for Metabolic Reconstructions
In Silico Methods for Strain Optimization
Once the metabolic network reconstruction has been manually curated, constraint-based modeling approaches can then be performed to characterize aspects of the organism's metabolism. One commonly used approach, flux balance analysis (FBA), can be used to predict the fluxes of metabolites through the metabolic network when optimized toward a biologically relevant objective, such as cell growth or the production of a metabolite of interest. FBA simulations can be performed using software tools like the COBRA Toolbox, a freely available software package that runs in MATLAB, and the BioMet Toolbox, an online resource for integrating high-throughput data for systems analysis. 105,106
To perform FBA, the metabolic network reconstruction must first be converted into a mathematical framework in the form of a stoichiometric matrix,
Because the resulting system of equations is underdetermined (ie, there are typically more reactions than metabolites in a reconstruction), FBA converts this system to a linear programming problem by optimizing for a particular flux called the objective function. Examples of objective functions include biomass production as an estimate for cell growth yield or the production of a metabolite of commercial interest. Additional constraints, such as enzyme capacities, maximum uptake, secretion rates and thermodynamic constraints, can be placed on the system to further narrow the solution space of feasible flux distributions. 108 Using these constraints and the specified objective function, FBA then calculates a distribution of fluxes through the metabolic network corresponding to the optimal value of the objective.
Since its introduction, several extensions have been made to FBA that have potential applications for in silico metabolic engineering. For example, in silico gene knockout simulations can be performed by taking advantage of the GPR relationship defined in the reconstruction to predict the effect that a model gene deletion or a combination of gene deletions will have on either biomass production or on the yield of a metabolite of interest. 109 However, given the size of genome-scale metabolic network reconstructions, the amount of data generated from in silico gene deletion combination simulations has proven to be challenging to manage. 110 To combat the immense volume of combinatorial data, several frameworks have been proposed to guide in silico gene knockout studies in a systematic and biologically relevant manner. OptKnock, for instance, is structured as a two-part optimization problem that evaluates the effect of gene knockouts to maximize both the overproduction of a metabolite of interest as well as the biomass. 111 Several extensions to OptKnock such as OptGene, OptStrain, OptORF, and OptReg have been proposed to help further guide in silico metabolic engineering gene deletion simulations. 112 –115 OptReg, for instance, extends OptKnock by allowing for gene up- and/or down-regulation, enabling the development of a broader range of potential metabolic engineering strategies.
Additionally, several FBA-based algorithms have been developed to depict more clearly the objective function that best characterizes the metabolism of mutant organisms. Studies have suggested that, unlike the wild-type phenotype, mutant organisms that have not been exposed to long-term evolutionary pressure do not operate in an optimized manner in regards to biomass production. Rather, in response to a genetic perturbation, the mutant tries to limit the difference in flux distribution between itself and the wild-type phenotype. In an effort to capture this altered objective function, algorithms like Minimization Of Metabolic Adjustment (MOMA) and Regulatory On/Off Minimization (ROOM) have been developed to minimize the predicted redistribution of metabolic fluxes in mutant phenotypes after a gene knockout by applying a minimal distance metric that limits the overall change in flux distribution compared to the wild type. 116,117
Further aiding in strain design, several methods like flux variability analysis (FVA) and flux coupling analysis (FCA) have been developed to study the structural and topological properties of metabolic networks. FVA identifies the range of possible fluxes for each reaction in a network given a certain objective function value. 118 FCA identifies pairs of metabolic fluxes in the network that are coupled together directionally, partially, or fully, as well as blocked reactions that are unable to carry a flux under certain conditions. 118 –120
Finally, genome-scale metabolic network reconstructions are an excellent platform for the integration of high-throughput data, allowing for the creation of context-specific computational models that have the potential to aid in identifying mutant strains suitable for the overproduction of a metabolite of commercial interest. As such, several algorithms have been developed for the integration of a variety of “omics” data, with special interest given to the integration of expression data due to the wealth of transcriptomic data currently available. One such method, Gene Inactivity Moderated by Metabolism and Expression (GIMME), employs a user-defined threshold to enable the methodical removal of reactions below the threshold. 121 Additional methods include the Integrative Metabolic Analysis Tool (IMAT), which creates a functioning model for fluxes based on mRNA transcript levels without relying on a user-specified objective function, and Metabolic Adjustment by Differential Expression (MADE), which uses the statistical significance of changes in gene expression measurements to turn model genes on and off. 122–123 Existing methods and integration techniques are improving continually. The current state of such methods, including E-flux and Probabilistic Regulation of Metabolism (PROM), has been reviewed by Blazier et al. 124
The use of FBA, FVA, and in silico gene knockout simulations performed on algal metabolic network reconstructions is discussed in further detail in the following section. Other algorithms, such as the computationally efficient combinatorial gene knockout algorithms, and the methods for omics data integration, have yet to be used with algae reconstructions, primarily because genome-scale algae metabolic network reconstruction development and analysis is still in its infancy. 125 Nevertheless, these algorithms have much potential to aid metabolic engineering strategies in genome-scale metabolic network reconstructions and provide biological insight into algal species.
Algal Metabolic Network Reconstructions
Although the genomes of at least 20 different algal species have been sequenced (Fig. 1C;
Sequenced Genomes and Existing Metabolic Models for Algae a
Compartments for each reconstruction are listed as follows: B. braunii was not compartmentalized; primary network of C. reinhardtii includes cytosol, mitochondria, and chloroplast; iAM303 includes cytoplasm, endoplasmic reticulum, extracellular, Golgi apparatus, lysosome, mitochondria, nucleus, peroxisome, and plasma membrane; C. reinhardtii thermodynamic model is not compartmentalized; iRC1080 includes mitochondria, glyoxysome, extracellular space, nucleus, Golgi apparatus, flagellum, eyespot, thylakoid, chloroplast, and cytosol; AlgaGEM is divided into cytosol, mitochondria, plastid, and microbody (peroxisome). Both O. lucimarinus and O. tauri models were not compartmentalized.
Prior to reconstructing the full genome-scale metabolic network of C. reinhardtii, several initial efforts focused on the reconstruction of the central metabolic network, with each successive iteration accounting for additional properties of the algal species. The first published model for algae focused on the central metabolism of C. reinhardtii and included 458 intracellular metabolites and 484 metabolic reactions, accounting for fatty acid, amino acid, and nucleotide synthesis as well as glycolysis and the citric acid cycle. 126 This model accounted for reaction and metabolite localization in three compartments: the cytosol, the mitochondria, and the chloroplast. Using flux balance analysis, the intracellular metabolic fluxes were predicted for C. reinhardtii growth under three different conditions: autotrophic, heterotrophic, and mixotrophic. Each of these three growth conditions was modeled by modifying the limits on CO2 and acetate uptake reactions as well as setting a constraint for the amount of light absorbed by the system.
Using an iterative methodology that combined bioinformatics and experimental techniques, a second central metabolic network reconstruction, iAM303, was generated with 259 reactions and 467 metabolites across several cellular compartments including the cytosol, mitochondria, chloroplast, glyoxysome, and flagellum. 127 In order to validate the model, the results from FBA after optimization of the network for either biomass or adenosine triphosphate (ATP) production were compared to experimental values found in the literature. Additionally, in silico gene-knockout simulations and FVA were used to propose genetic engineering strategies for increased hydrogen production. A non-compartmentalized model of the primary metabolism was also created by incorporating thermodynamic parameters with constraint-based modeling techniques to determine the extent of light-driven respiration. 128 By constructing a two-step objective function, including both the growth rate of cells and the photon uptake rate, this study allowed for simulation of biomass growth under low light conditions, revealing interactions between respiratory activity and photosynthesis in C. reinhardtii.
More recently, two genome-scale metabolic network reconstructions have been generated for C. reinhardtii. One such reconstruction, iRC1080, accounts for the function of 1,068 metabolites and 2,190 reactions localized across 10 compartments, and was the first metabolic network reconstruction to account for photon absorption and growth simulations quantitatively under various light sources. 129 Using the C. reinhardtii central metabolism reconstruction iAM303 as a starting point, Chang et al. added reactions on a pathway-by-pathway basis using more than 250 publications. iRC1080 also significantly expanded the number of lipid metabolic pathways over preceding metabolic reconstructions, enabling the application of in silico metabolic engineering strategies aimed at enhancing lipid production. In order to validate the model, in silico gene knockout simulations were performed and subsequently compared to published data on mutant C. reinhardtii phenotypes. FBA and FVA were subsequently used to simulate adaptations in metabolic response to changes in light influx for contribution of metabolic pathways to biomass production, including lipid synthesis and protein production. 132
AlgaGEM, another C. reinhardtii genome-scale metabolic network reconstruction, accounts for the function of 1,869 metabolites and 1,725 metabolic reactions across several cellular compartments such as the cytoplasm, the mitochondrion, the plastid, and the microbody. Compartmentalization was determined from published literature or by comparing homologs to a metabolic model of the plant species A. thaliana. To identify gaps in the model, FBA was used to evaluate the model's ability to produce major biomass components under autotrophic, heterotrophic, and mixotrophic conditions. AlgaGEM was subsequently used to predict metabolic targets for the increased production of H2 by performing in silico gene knockout simulations on the network. 130
In addition to those curated for C. reinhardtii, genome-scale metabolic network reconstructions have been generated for the green algae O. lucimarinus and O. tauri. Ostreococcus species are prevalent as microalgae and are ideal organisms to study because of their simplicity and phylogenetic relationship as early-diverging green plants. 131 Furthermore, O. tauri is becoming popular as a model organism for metabolic studies as it has the smallest eukaryotic genome and an unusually strong adaptation to nutrient stress, demonstrated in part by its ability to respond to low nitrogen conditions—suggesting a fast global metabolic response. 133 To generate genome-scale metabolic network reconstructions for both O. lucimarinus and O. tauri, draft networks were first reconstructed for each species using the KEGG database. Subsequently, a sophisticated FBA-based gap-filling algorithm that took into account phylogenetic distance among species was applied to the two draft reconstructions, resulting in more complete and accurate networks. Additionally, this algorithm highlighted gaps in the annotation of genes for both Ostreococcus species.
Finally, in the absence of a fully sequenced genome, a model for the organism B. braunii, a high oil-producing algal strain, was constructed using transcriptomic data and various databases including KEGG, MetaCyc, and Reactome. 17 The ability of B. braunii to produce large volumes of terpenoid liquid hydrocarbons, which are similar to crude fossil fuels, motivated the study of the genes and pathways involved in terpene production. Using a large-scale next-generation sequence read dataset, the Department of Energy Joint Genome Institute produced a metabolic network through various annotation methods, including the B. braunii Showa web-based annotation tool, FrameDP, and the NetStart 1.0 Server, to identify genes and pathways key for the production of terpene compounds and precursors. As a result of this study, an annotated transcriptomic database for B. braunii was made publicly available online, paving the way for future in silico metabolic engineering analyses such as gene knockout simulations to optimize the production of terpenoid liquid hydrocarbons.
Simulations for the majority of C. reinhardtii models, including iAM303, iRC1080, and AlgaGEM, were completed with the COBRA toolbox implemented in MATLAB, freely available from the openCOBRA project (
Industrial Applications of Biotechnology
A key to making algae-to-energy systems viable and sustainable in the long run will be to tie emerging metabolic network models with the large-scale systems drivers that will determine the industrial relevance of these processes. Ultimately, the technical and economic viability of a scaled up algae-based biotechnology sector, especially for products to be produced in large quantities such as fuels, will depend largely on the availability of land, water, nutrients, and CO2, as well as markets for byproducts, among other constraints. 135 A great majority of existing algae biotechnology research has taken for granted the supply chains and large-scale systems that will need to be put in place to support such an industry at relevant scales. 136 This is starting to change as life cycle analysis and other tools from the emerging field of industrial ecology are being used to try and understand more about ancillary processes. 137 By evaluating biological viability and industrial sustainability in unison, in silico metabolic engineering and industrial ecology can be used to optimize parameters in a way that maximizes the potential of industrial algae processes.
A number of companies currently produce compounds from algae including ethanol, hydrogen food colorants, and biofuel precursors. 138 Several chemical companies, such as Synthetic Genomics (La Jolla, CA) and Sapphire Energy (San Diego, CA), have employed metabolic engineering strategies to improve fuel yields from algae. 139 Solazyme (San Francisco, CA) has produced and begun commercializing biobased fuels from heterotrophic algae, although precise synthetic biology and metabolic engineering strategies have remained proprietary. 140 Other algae production companies and their methods for biofuel conversion have been previously described. 141 A large number of startup biotechnology companies are also working to leverage computational metabolic methods in the production of cosmetics, pharmaceuticals, and specialty chemicals; Rosetta Green (Rehovot, Israel), for example, is working toward the successful production of human growth hormone from algae.
Conclusions
Efforts to deploy metabolic optimization strategies are limited primarily by the complexity inherent in understanding biological systems. To overcome this complexity, efforts to leverage metabolic network models have tended to focus on single factor optimization. In the context of algae-biofuel production, for example, there has been a heavy focus on the maximization of lipid yields. 14 Empirical data from years of laboratory work suggest that very high lipid yields (>50% mass) are possible, but that these yields also result in lower growth rates. 142 If the total lipid productivity is higher, even though the cell growth rate is lower, then such efforts to maximize lipid production would be worthwhile. While tradeoff analysis is relatively simple on a pathway-by-pathway basis, it becomes challenging for increasingly complex metabolic systems. Metabolic network reconstructions along with in silico analyses such as FVA can be used to aid in the tradeoff assessment for both simple and complex metabolic systems.
A great opportunity exists to couple metabolic engineering with regional materials flow analysis. Material flow analysis is a quantitative tool for measuring material stocks and flows at the scale of regions or nations. 143 Continuing on with the algae-biofuel example, this coupling would enable algae farmers to select strains that are both regionally appropriate and capable of achieving maximum profits under current market conditions. Knowing how water composition and average temperatures affect growth rates in a variety of strains could help an algae farmer select a family of algae that would be the most viable strains for their site. The developer could additionally refine strain selection by considering which byproducts would be most valuable under prevailing market conditions. Obtaining this information using traditional laboratory approaches would be immensely expensive and limited because it would not allow for the manipulation of algae cells for some generally desirable traits; however, as genome-scale metabolic network reconstructions become more encompassing, they may be used to address such concerns as optimal strain selection.
Interestingly, the computational structure of most tools in industrial ecology, including life cycle assessment and material flow analysis, has parallels with metabolic network reconstructions. These models are generally comprised of large systems of equations that describe material flows resulting from deliberate human activity in the economy or via indirect or undesirable pathways in the form of pollution. 144 These systems of equations can be manipulated depending on the system boundaries or functional unit of interest. 145 Over the past decade, methods for obtaining data that are temporally and spatially specific for material and energy flows have resulted in several large commercial databases that greatly facilitate the development of life cycle and material flow models. 146 The similarities between the numerical methods used in both problems suggest that they could one day be integrated to yield algae strain selection tools that span huge spatial scales.
There are certain phenotypes that will be desirable for all industrially relevant algae. Most importantly, species that maximize available CO2 will be prized given how difficult it will be to set up a CO2 supply chain to deliver CO2 to large-scale algae cultivation facilities. Most analyses have tended to assume that CO2 will be obtained from a large point source, such as a coal or natural gas-burning power plant. 147 Coal-fired plants are ubiquitous and their flue gas contains high levels (∼12%) of CO2. The flue gas tends to be dirty, however, containing significant particulate matter such as SO2 and NOx, which can impact the pH of pond water if it is not remediated first. Flue gas from natural gas power plants is much cleaner but it has lower levels of CO2 and, consequently, the costs of separating out the CO2 are higher. Given how involved it will likely be to supply large industrial facilities with CO2, designing algae to use CO2 more efficiently would improve the efficiency and bottom line of the company. 148 In silico gene knockout simulations could be performed on metabolic network reconstructions using algorithms like OptKnock and MOMA to aid in the design process of mutant algae that more efficiently metabolize CO2.
Other important phenotypic characteristics include the algae species' ability to tolerate high salinity growth media; produce compounds that would enable them to outcompete invasive species; remain neutrally buoyant in growth ponds; or be capable of autoflocculation. 149 These characteristics, which can be understood in part in the context of metabolic network models, will have important ramifications on the engineering processes necessary to use algae for biofuels production. Employing these metabolic network models will help ensure that genetic modifications are being carried out in the most rational and efficient way possible, and that the most appropriate tools are being developed for the burgeoning industry.
Footnotes
Author Disclosure Statement
No competing financial interests exist.