
Abstract
Thermobifida fusca Cel6A is an endoglucanase with relatively high activity on crystalline cellulose. Trp 231 is a substrate-binding residue in the −2 sub-site that is within the active-site cleft. Mutating Trp 231 to Ala, Asp, or His causes significant decreases in activity on both oligosaccharides and cellulose. The largest decreases in activity are seen on short oligosaccharides, which is clearly due to altered oligosaccharide binding, as shown by docking studies. The mutation causes a significant drop in the production of soluble products from crystalline cellulose, with little change in the production of insoluble reducing ends. This result is due to weaker rebinding of the mutant enzyme to cellulose after hydrolysis before it completely dissociates, while the initial binding of the mutant catalytic domain is only slightly reduced. The Asp mutant enzyme had a much smaller increase in activity with increased temperature than the wild type, but it is not clear why this occurs. However, this mutant enzyme also showed weaker binding to cellulose at higher temperatures, presumably because of the role of Trp 231 in substrate binding; this might explain the temperature effect on the mutant enzyme.
Introduction
Understanding how cellulases digest crystalline cellulose is important for producing alternative energy sources that are environmentally friendly and renewable. Cellulases may play a critical role in biofuel production because of their ability to hydrolyze recalcitrant cellulose into glucose, which can then be fermented to produce biofuels. 1 Although cellulases have been extensively studied, attempts to engineer them for increased activity have not been very successful, due to a lack of information about their rate-limiting steps. A better understanding of these enzymes would allow production of more reactive cellulases for specific pretreated biomass substrates. This would reduce the cost of cellulose hydrolysis, making the production of fuels and chemicals from biomass more economical. 2
Cellulose is a homopolymer that consists of D-glucose units connected by β-1–4 glycosidic linkages. This polymer contains both inter- and intramolecular hydrogen bonding networks that cause cellulose to be recalcitrant to enzymatic degradation. 3 Although many hypotheses have been proposed, we still do not know how a cellulase disrupts a microfibril and productively binds to a single cellulose chain.
Thermobifida fusca is a thermophilic aerobic soil bacterium that produces six cellulases that act synergistically to hydrolyze crystalline cellulose. 4 One of these cellulases, Cel6A, is a modular endocellulase consisting of a catalytic domain (CD)—residues 38–320—and a carbohydrate-binding domain (CBD)—residues 343–441—joined by a flexible linker (residues 321–342) (Fig. 1). 5 It produces cellobiose (G2), cellotriose (G3), and glucose (G1) from cellulose. 6,7 The immediate and constant production of soluble oligosaccharides suggests that Cel6A makes several cleavages during each productive binding event. 4,8 The structure of the Cel6A catalytic domain was determined by x-ray crystallography, and it has a modified α/β barrel protein fold. 9 Cel6A uses an inverting mechanism involving a catalytic acid, Asp117, and a catalytic base, probably involving Asp79 and Ser84. 10,11 Its catalytic domain has five glucose-binding sub-sites: −3 to +2; W231 is located in the −2 sub-site, which has been shown to require glucosyl occupation for hydrolysis to occur in other family 6 cellulases. 12,13 To understand the mechanism of cellulase active-site binding and catalysis, mutagenic studies have been used to study Cel6A to elucidate the role of key residues. 14 Mutating Cel6A W231 to Asp, His, or Ala causes dramatic changes in its activity and this study examines the role of W231 in substrate binding and activity.

Model of Cel6Acd, with Tryptophan 231 highlighted in black and cellotetraose in the active site cleft.
Materials and Methods
Protein Purification
Cel6A plasmids (pet26b+) were isolated from Escherichia coli DH5α, and strains were grown in Luria-Bertani (LB) broth with 60 μg/mL kanamycin and stored with 10% glycerol. All proteins were expressed in E.coli BL-21(DE3). Starter cultures (33 mL) were grown in LB overnight and inoculated into 1 L M9 medium. After growth for 5 h at 30°C to A600=0.8, cultures were induced with 0.5 mM isopropyl thio-β-D-galactoside (IPTG, Sigma-Aldrich, St. Louis, MO) and harvested after 4 more hours of incubation. 15 Enzymes were purified by chromatography on P-Sepharose, Q-Sepharose, and hydroxyapetite columns (Sigma-Aldrich, St. Louis, MO) as reported previously. 16 Wild type (WT) Cel6Acd was produced by papain digestion of WT Cel6A. 16
Substrates
Bacterial microcrystalline cellulose (BMCC) from Acetobacter xylinum (Monsanto Cellulon, San Diego, CA) was prepared by centrifugation, washing with distilled/deionized water three times at 4°C, and suspension in 50 mM NaOAc pH 5.5 at a concentration of 5 mg/mL. Carboxymethyl cellulose (CMC, Sigma-Adrich) was dissolved to a concentration of 2% in 50 mM NaOAc pH 5.5 at 4°C. Swollen cellulose was produced by treating Sigmacel (Sigma-Aldrich) with concentrated phosphoric acid as described previously. 17 All substrates were stored in 0.04% azide to prevent microbial growth. In assays using BMCC and swollen cellulose (SWC), 1 mg of substrate was used per reaction. For filter paper, a standard hole punch was used to create disks from Whatman #1 (Sigma-Aldrich) with an average mass of 8 mg, while for CMC, the final assay concentration was set at 1%.
Mutagenesis and DNA Manipulations
Mutation of Cel6A to produce W231D, H or A, was carried out on the pet26b+vector plasmid with the QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, La Jolla, CA) following the manufacturer's protocol. Cel6AW231Dcd was created by a two-base-pair mutation of G287 to form a stop codon, deleting the linker and the CBM. Lasergene software (DNAStar, Madison, WI) was used for analysis of Cel6A mutations as well as calculations of molecular weight, isoelectric point, and extinction coefficents.
Activity Assays
Enzyme concentrations were determined at A280 using a Nanodrop Spectrometer (Thermo Scientific, Wilmington, DE) using an extinction coefficient calculated from the sequence in Lasergene: Cel6A-WT:81.7k L/mol/cm; Cel6A-W231D:76.1k L/mol/cm; Cel6Acd-WT:52.4k L/mol/cm; and Cel6Acd-W231D:57.9k L/mol/cm, with molecular weights of 42.9kDa, 42.8 kDa, 30.3 kDa, and 30.4 kDa respectively. Activity was calculated in the units of μM reducing ends/time/μg enzyme when 0.2-μM reducing ends were produced.
DNS assay
The activity of the enzymes was determined by the measurement of the reducing sugars calibrated with a glucose standard curve using 3,5-dinitrosalicylic acid reagent (DNS, Sigma-Aldrich), as previously described. 4 BMCC and filter paper were assayed overnight with enzyme concentrations of 23–465 pmol per reaction. SWC and CMC were assayed over 1 h with 4.6–37.2 pmol enzymes per reaction. All assays were run at a final volume of 0.40 mL in 50 mM NaOAc buffer (pH 5.5).
DNS insoluble assays
To determine the amount of reducing ends present in the insoluble fraction of substrate, 0.8 μg enzyme was reacted with 1 mg of BMCC (total reaction volume=400 μL in 50 mM NaOAc buffer, pH 5.5). After incubating overnight at 50°C, the samples were boiled for 10 min to inactivate enzymes. The insoluble substrate was washed three times by centrifuging at 16,060 G, with removal of 300 μL of supernatant, and replacement with 300 μL of 50 mM NaOAc buffer. Reducing sugars were assayed using DNS reagent, as described above.
Oligosaccharide assays
Enzyme and oligosaccharide stocks at 2x reaction concentration were prepared and pre-incubated at 50°C for 30 min. Cellotetraose (G4), cellopentaose (G5), and cellohexaose (G6) (Megazyme, Wicklow, Ireland) were assayed in a final reaction volume of 600 μL with an enzyme and oligosaccharide concentration of 20 nM and 100 μM, respectively. Samples were incubated for 10 min at 50°C and immediately boiled for 10 min to deactivate the enzymes. Cellotriose G3 was incubated for 24 h due to low activity. The reactions were analyzed by high-performance liquid chromatography (HPLC) (Shimadzu, Columbia, MD) using a refractive index detector. The injection volume of each sample was 50 μL. A flow rate of 0.60 mL/min was used through an HPX-87P Bio-Rad Aminex column (Bio-Rad, Hercules, CA) at 84°C using MilliQ water (Millipore, Billerica, MA) as the mobile phase. Glucose, cellobiose, G3, and G4 were used as standards, and the HPLC chromatograms were analyzed via integration using Origin software (OriginLab, Northampton, MA).
BMCC/SWC/CMC assays (HPLC)
For SWC and CMC, 4.6–37.2 pmol of enzymes were used in a final volume of 600 μL containing 1 mg of SWC or CMC. For BMCC, 23–465 pmol of enzyme was used with the same conditions. All samples were filtered through Costar Spin-X (Corning, Corning, NY) columns and then analyzed by HPLC as described above.
Thermal Stability
Cel6A, Cel6A-W231D at 92 nM, and CMC 2% w/v were pre-incubated at 60°C for 1 h. CMC and enzyme were combined to a final reaction volume of 400μL, incubated at 50°C for 1 h, and analyzed by DNS, as described above. Circular dichroism (CD)-spectra were performed with an Aviv Circular Dichroism Spectrophotometer (Lakewood, NJ) using a 0.2-cm quartz cuvette, protein concentrations of 0.05 mg/mL, and temperatures of 30–60°C.
Binding Assays
Enzyme concentrations at 15 pM were assayed with 0.25 mg of BMCC in a final volume of 100 μL at 4°C and 60°C in low-bind tubes for 1 h. Samples were then centrifuged for 5 min at 13,000 rpm. Free enzyme in solution was detected at A280 using a NanoDrop Spectrometer (NanoDrop, Wilmington, DE), and bound enzyme was calculated by subtracting enzyme and substrate controls.
Computational Binding Affinities and Docking
The software Autodock Vina was used in conjunction with MGLTools, SwissPDB, and Pymol to perform surface docking of the oligosaccharide within the active-site cleft of Cel6A and Cel6AW231D as recommended. 18 The Cel6A crystal structure used in docking experiments was TML1.pdb from the RCSB protein databank.
Results
Activity Assays
The product distributions of Cel6A WT and the W231D enzyme were determined after overnight hydrolysis of BMCC. Cel6A produced 6% G1, 81% G2, and 12.5% G3, while W231D retained 31% of the activity of WT and produced 0.8% G1, 58.7% G2, 40% G3, and 0.53% G4. Cel6Acd and W231Dcd had 45% and 47% lower activity than the intact enzymes on BMCC, respectively. The effect of temperature on SWC activity was determined, and the results show that increasing temperature had a greater effect on Cel6A than on the Asp mutant enzyme (Fig. 2). It is interesting that at 60°C the activity of the W231D enzyme decreased on CMC. To determine whether the decrease in activity was caused by denaturation, thermal stability was assayed by pre-incubating the enzymes at higher temperatures; neither enzyme showed reduced activity, even after a 60°C incubation. CD spectra were also performed at several temperatures, and no conformational changes were observed in the mutant enzyme.
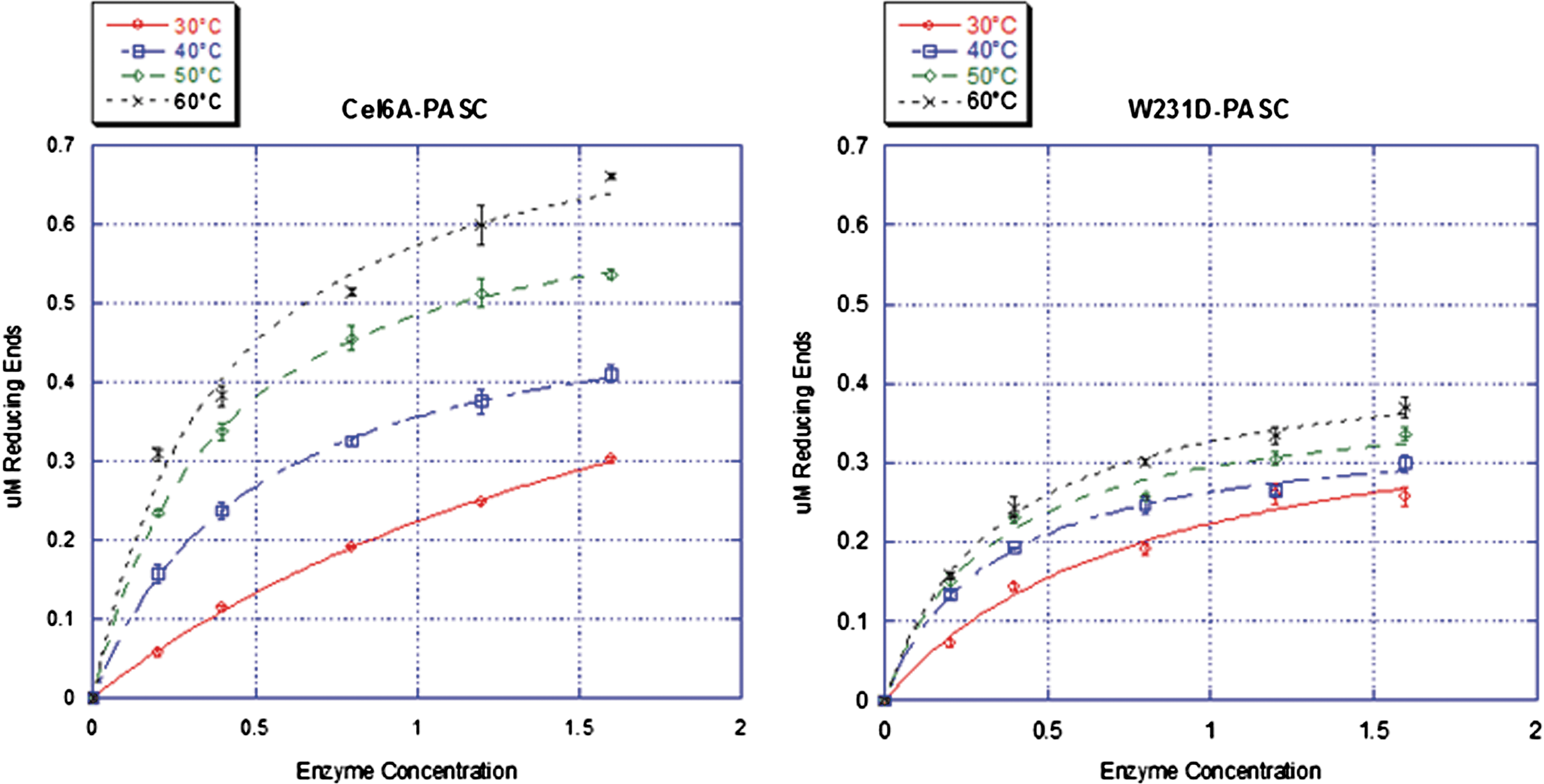
Activity assays of Cel6A-WT and W231D on BMCC; the difference of activity is more apparent at higher temperatures. Color images available online at
To investigate further the effects of the mutation on BMCC activity, the amounts of soluble and insoluble sugars produced from BMCC were determined for both enzymes. The ratio was 2.8 for Cel6A and 1.0 for the W231D enzyme (Fig. 3).
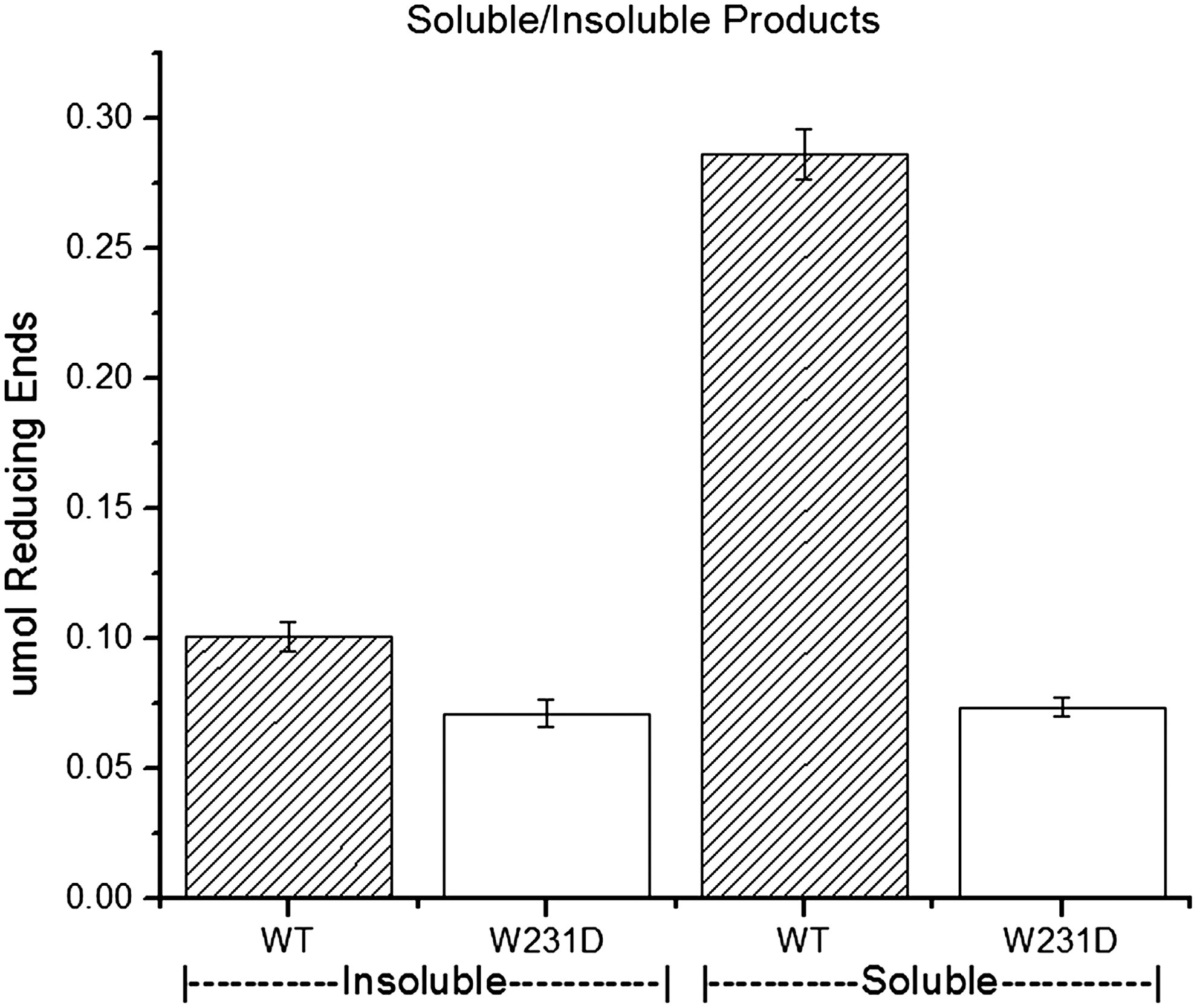
DNS assay of soluble to insoluble products produced by Cel6A-W231D and WT shows a decrease in soluble products produced by the mutant enzyme.
Cel6A also was mutated to W231H and W231A to demonstrate that the activity change was caused by the loss of Trp231 rather than the presence of Asp, and both these mutant enzymes had 76% lower activity than WT, similar to the W231 Asp mutant enzyme. HPLC analysis of their products also showed an increase in G3 and G4 relative to G2.
Binding Assays
Binding assays were performed to determine if altered substrate binding was associated with the decrease in the mutant enzyme activity. The W231D enzyme has the same binding activity to BMCC as WT, with 77% binding at 4°C—the temperature showing maximum binding. At 60°C, WT binding was reduced to 70% and W231D was reduced to 48% (Fig. 4). No binding was observed with either enzyme when the CBM2 was removed.
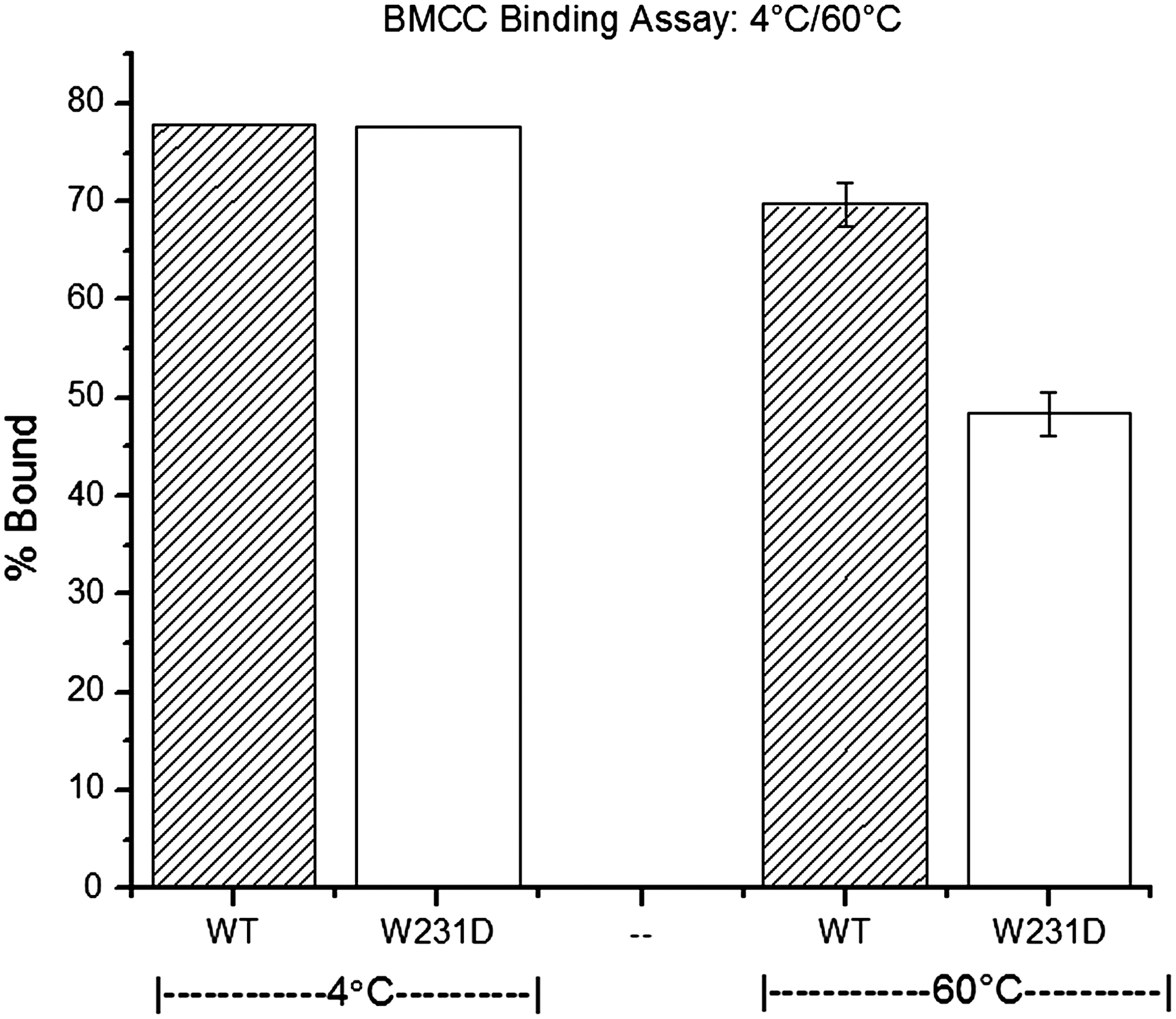
Temperature-dependent binding is observed for the WT and mutant enzymes. Lower binding is a plausible explanation for lower activity at higher temperatures.
Oligosaccharide Assays
The change in product distribution (i.e., the increase in G3 and the presence of G4) led us to assay oligosaccharide activity. Hydrolysis of oligosaccharides by the W231D enzyme was less than WT for G4, G5, and G6 by 99%, 74%, and 18%, respectively, and G3 was not hydrolyzed. The product distribution for WT G6 hydrolysis was 62% G2, 32% G3, and 6% G4, while W231D hydrolysis of G6 yielded 18% G2, 71% G3, and 11% G4. The percentage of each oligosaccharide resulting from hydrolysis events was calculated based on the total amount of bonds broken by WT (Fig. 5).
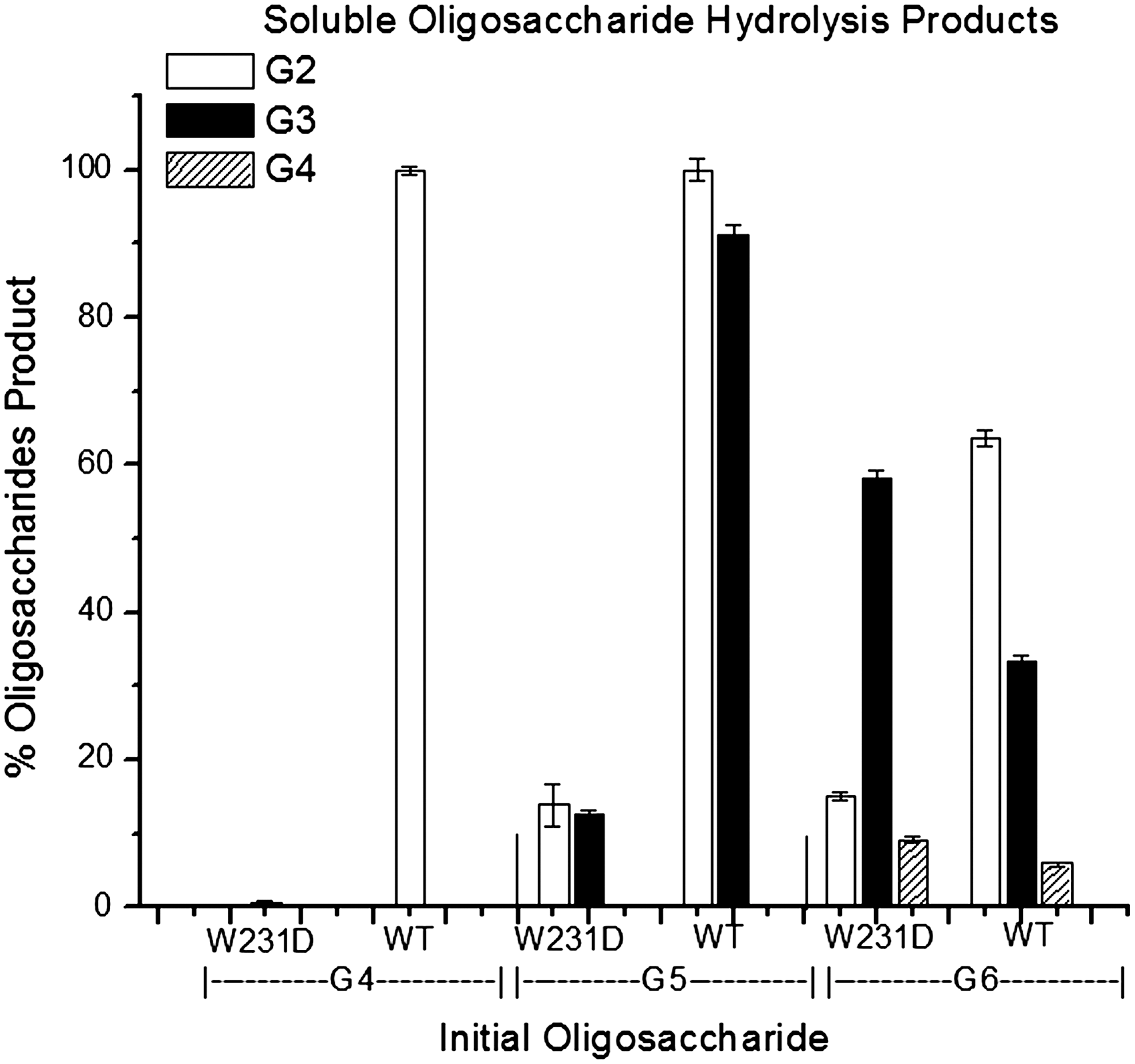
Hydrolysis by Cel6A and the mutant enzyme shows that the mutant enzyme retains dramatically more activity on longer oligosaccharides and the product distribution from cellulose is changed by the mutation. The inability of W231D to hydrolyze G4 effectively suggests a processive element in Cel6A due to the higher ratio of G2 over G4.
Auto-Dock Simulation
The W231D, W231H, and W231A enzymes all had different binding sites for G4 than did the WT enzyme in docking simulations. G4 binds to WT in the expected +2 to −2 sub-sites. Changing W into another aromatic residue, H, shifted the binding into the +1 to −3 sub-sites and when the aromatic residue was removed, binding shifted to the +4, to +1 sub-sites in both enzymes explaining their low activity (Fig. 6).
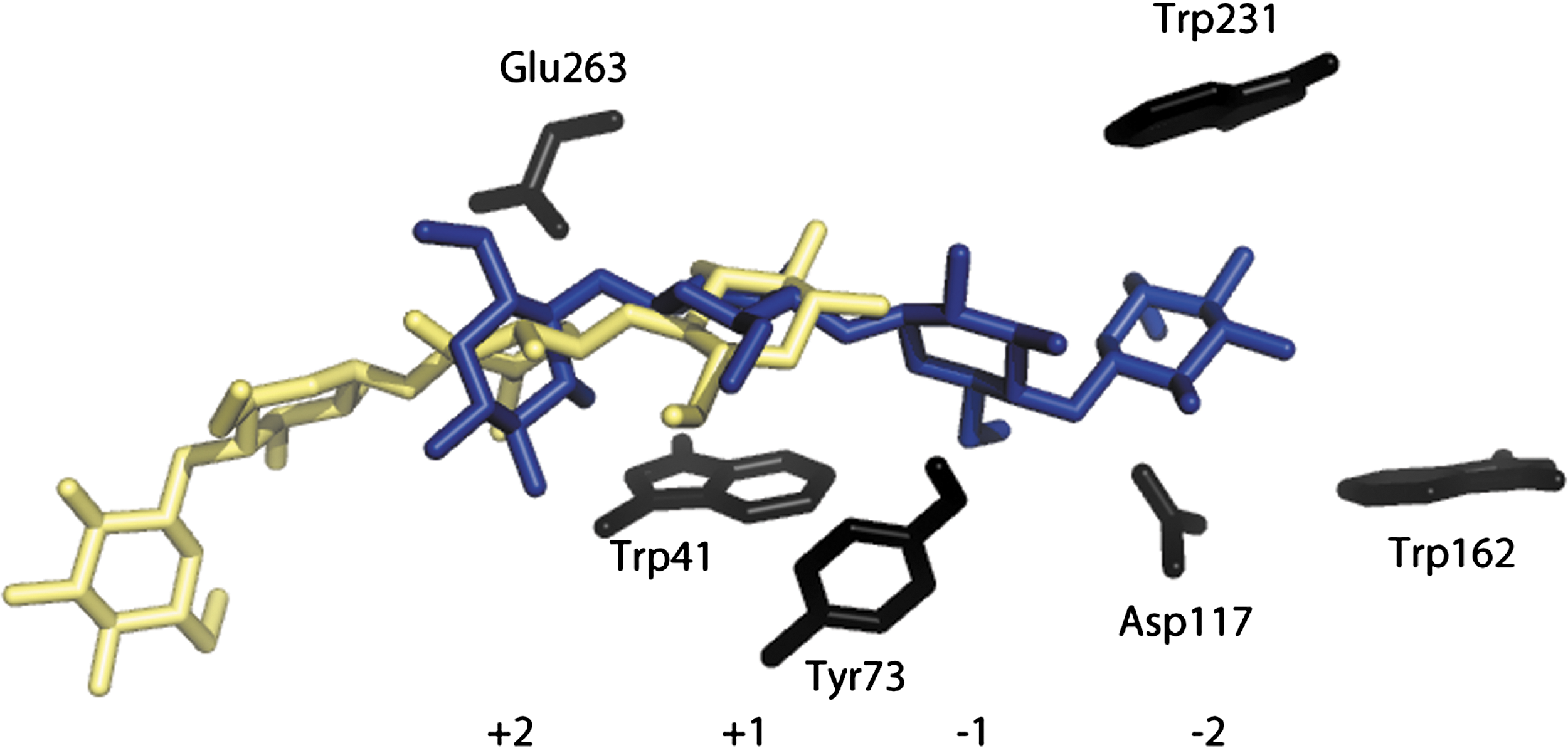
Cel6A binding of G4 in the active site. Trp 231 is located in the −2 sub-site. Binding of G4 in the catalytic site of the A mutant enzyme is shown on left, with WT binding on right. Color images available online at
Discussion
The distribution of products from BMCC hydrolysis by Cel6A WT and the W231D enzyme was very different; 1-h incubation produced G3, G2, and G1 for WT and G4, G3, and G2 for W231D. The different products can be explained if the WT enzyme produced all the soluble oligosaccharides, but cleaved the longer ones too quickly for them to be detected. A 10-min incubation with each oligosaccharide showed that WT cleaved at least 80% of G4, G5, and G6, while the W231D enzyme activity on these oligosaccharides was much lower—only 1% of WT activity on G4. An interesting result occurred with G6 hydrolysis, in which two modes are possible: the production of G2 and G4 or two G3s. Because the W231D enzyme hydrolyzes G4 poorly, it should make equal amounts of G4 and G2; however, it gave a 40% excess of G2 over G4 (Fig. 5). This nonequivalence may be explained by W231D binding to G6, producing the G2 and G4 products and being pseudo-processive on the G4 to produce two G2s. The W231D enzyme hydrolyzed G5 more slowly than WT. The nonequivalence of G2 and G3 in the WT hydrolysate is due to a small amount of G3 hydrolysis, which does not occur with the mutant enzyme.
The W231H and W231A enzymes exhibited a similar decrease in BMCC activity (76%) and similar product distribution as the W231D enzyme, showing that it is loss of the W residue—not the presence of D—that changes activity.
The activity of the WT and mutant enzymes on BMCC were studied further by measuring the amounts of insoluble and soluble reducing sugars that they produced. If Cel6A acted by making a single cleavage each time it attaches to cellulose, one would expect very little soluble product at early time points. The ratio of soluble/insoluble product was determined to be 2.8 for WT and 1.0 for the W231D enzyme (Fig. 3). Assuming that insoluble reducing ends produce a comparable signal to soluble reducing ends in the DNS assay, these data show that the W231D enzyme maintains its ability to catalyze glycosidic cleavages on the insoluble surface but produces fewer soluble products, implying that it is unable to make all of the additional cleavages made by WT enzyme, which appears to produce four cleavages per binding event.
The molecular graphics program Auto-Dock Vina was used to predict the stable binding sites for oligosaccharides on the enzymes. The most stable binding conformation of G4 to WT is in the expected +2 to −2 sub-sites, whereas for the W231D and W231A enzymes binding was in the +1 to +4 sub-sites, explaining their low hydrolysis rates (Fig. 6). This is expected, as loss of the CH/π system provided by the tryptophan destabilizes the binding site. The W231H enzyme bound G4 into the +1 to −3 sub-sites. Docking shows a ring-stacking interaction between 231H and the −2 position. The W231H enzyme also produced G4, similar to the W231D enzyme but unlike WT in phosphoric acid swollen cellulose assays.
Activity assays were conducted between 30–60°C, and the change of temperature had a major effect on WT activity with an average increase of activity of 0.74 μmol reducing ends (RE)/min/μmol/10°C. Temperature change had much less effect on W231D enzyme activity with an average change of 0.24 μmol RE/min/μmol/10°C. At lower temperatures, the W231D enzyme is similar to WT (within .02 μmol RE/min/μmol/10°C) (Fig. 2). To determine whether this change in activity was caused by denaturation, CD-spectra were determined at 30–60°C, and no conformational changes were observed. Activity assays on enzymes pre-incubated for 1 hour before hydrolysis were performed and also showed no changes in activity. At 4°C, the W231D enzyme had similar binding to BMCC as WT. However, as the temperature was increased to 60°C, the W231D enzyme had 37% less binding than WT (Fig. 4). At higher temperatures, we propose that weakened hydrophobic interactions due to the loss of W231 caused increased dissociation. Although an increased dissociation usually causes lower activity, the data show that only short-chain oligosaccharide production is affected, but not insoluble cleavages, suggesting that one of the limiting steps for Cel6A soluble oligosaccharide production is interaction with enough binding sites to stabilize cellulose in the active-site cleft. The observation that the rate of insoluble end production is not lowered by the mutation may be attributable to the existence of residues in or on the active-site cleft that move a cellulose chain into the active site, resulting in its initial cleavage, as these residues should not be changed by the studied mutation.