
Abstract
Herein, the probiotic potential of Blautia producta was investigated by whole-genome analysis. The genome assembly leads were successful at 100% completeness with 3.8% contamination. The leads comprised 120 contigs spanning 5,965,788 bp, with an average gene cluster of 45.91% and a high N50 value of 142,259 bp, indicating excellent assembly quality. A total of 215 subsystems related to metabolism, protein processing, and stress response were identified. Moreover, 4630 encodes nearly 64 functional proteins involved in transcription, carbohydrate transport, signal transduction, and defense. Kyoto Encyclopedia of Genes and Genomes and Clusters of Orthologous Group analyses also revealed pathways involved in nutrient transport, protein synthesis, and quorum sensing. The two unique gene clusters identified, the nonribosomal peptide synthetase and ranthipeptide clusters, are known for their antimicrobial and signaling activities. Furthermore, B. producta showed no resistance to 116 tested antimicrobial agents and had low (42.7%) human pathogenicity. In contrast to other probiotics, B. producta also has similar genomic and functional attributes, which support various research and therapeutic applications.
Introduction
Over hundred trillion different commensal microbes inhabiting the human gastrointestinal tract (GIT) play a pivotal role in health benefits. 1,2 The microbiota, not only within bacteria but also closely associated with other microbial classes, remains relatively stable throughout the lifespan, resulting in self-regulatory responses within the host. 1,3 However, some antibiotics, immunosuppressive drugs, and radiation flux can alter the balance among the microbiota, which has serious health implications. However, during an altered state, these microbiota, more specifically bacterial flora, are known to restore host homeostasis. 4 Currently, there is significant knowledge on the effects of the gut microbiota on human health and on its functional role in maintaining gut health or overall homeostasis. 5 To overcome this, some probiotics are claimed to have health benefits when they are delivered adequately to the host GIT. However, these strains should adhere to and tolerate stress within the GIT lining, which then exerts an immune response to boost host homeostasis by synthesizing vitamins and certain amino acids. The probiotics not only closely associate with the other microbes in the GIT lining but also deliver antimicrobial and bacteriostatic molecules.
Blautia producta (genus: Blautia), a prominent member of the gut microbiota, is known for its various physiological roles in human health and disease control. 6 Moreover, B. producta is negatively correlated with type II diabetes and hypercholesterolemia, suggesting that B. producta may be a therapeutic target. 7 –10 Additionally, B. producta has hepatoprotective effects against lipopolysaccharide (LPS)-induced acute liver injury. B. producta is also known for its antioxidant activity and secondary metabolites, which further extend its integrated interest in pharmacology, comparative genomics, and metabolomics. 7 –12
Currently, thorough insight into the genome of probiotic strains is rapidly increasing to understand the relationships between disease alleviation states. Notably, whole-genome sequence (WGS) analysis of such probiotic strains will provide comprehensive insights into safety assessments, virulence genes, antibiotic resistance, and toxicity for food and pharma products. 12 This multistep evaluation of B. producta CB7BLD4 was needed for its probiotic leads due to the nonavailability of data at the genome level. Hence, this study aimed to identify the B. producta genomic landscape and functional annotations to facilitate its probiotic potential and to contribute to microbiome research for targeted interventions in the future.
Materials and Methods
DATA COLLECTION AND PREPROCESSING
The Illumina HiSeq 2000 paired-end sequences of the raw reads of the B. producta CB7BLD4 gene draft were obtained from the ENA Browser (https://www.ebi.ac.uk/ena/browser/view/SRX10204312). Further processing of the data was performed in Bacterial and Viral Bioinformatics Resource Center (www.bv-brc.org), 13,14 FastQC, 15 and Trim Galore (version 0.6.5_dev). In addition, SAMtools (version 1.17) 16 was used for processing aligned sequencing data in the Sequence Alignment Map/Binary Alignment Map (SAM/BAM) format, facilitating sorting, indexing, and filtering reads for subsequent analyses. Both the Unicycler (version 0.4.8) 17 and SPAdes (version 3.13.0) 18 assemblers were utilized for genome assembly, and the resulting assemblies were compared for quality assessment. Pilon (version 1.23) 19 was then applied for polishing both the assembled genome and correcting errors such as Single Nucleotide Polymorphism (SNPs) and indels. For the bandage plot, Bandage (version 0.8.1) was used for the identification and comparison of structural features of both genome assemblies separately.
GENOME ANNOTATION AND CAZyme PREDICTIONS
Following preprocessing, the assemblies generated by Unicycler and SPAdes were compared using Benchmarking Universal Single-Copy Orthologs (BUSCO) (version 5.5.0) 20,21 and Quality Assessment Tool for Genome Assembranes (QUAST) (version 5.2.0) 22 for quality assessment. For further downstream and interpretation, Prokka (version 1.14.6) 23 was used. Complete genome annotation was performed in Galaxy (https://usegalaxy.org). 24,25 In addition, the RAST tool kit (RASTtk)-Pathosystems Resource Integration Center (PATRIC) was used to ensure more accurate and comprehensive annotation results. The functional categories of the orthologous groups (OGs) were analyzed using egg-NOG Mapper (http://eggnog-mapper.embl.de/), 26 which integrates various annotation sources such as predicted protein names, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and Clusters of Orthologous Groups (COG) categories. Carbohydrate-active enzyme (CAZyme)-associated genes were identified in the B. producta genome using the dbCAN2 online server (https://bcb.unl.edu/dbCAN2/index.php) via a DIAMOND Basic Local Alignment Search Tool (BLAST) search of the CAZyme database (http://www.cazy.org).
Prediction of secondary metabolite-related gene clusters
Antibiotics and Secondary Metabolite Analysis Shell (antiSMASH) (v7.1.0) (https://antismash.secondarymetabolites.org/) 27 was used to identify and annotate biosynthesis-related gene clusters involved in secondary metabolite production in B. producta.
Testing for resistance, prophage genes, and predicting CRISPR–Cas
The antibiotic resistance profiles of B. fluorescens strains were identified via ResFinder (v4.5) (http://genepi.food.dtu.dk/resfinder), 28 PathogenFinder (v1.1) (https://cge.food.dtu.dk/services/PathogenFinder-1.1), and the Comprehensive Antibiotic Resistance Database (CARD) (https://card.mcmaster.ca/analyze/rgi). 29 For the prophage genes, the PHAGE Search Tool Enhanced Release (PHASTER) (https://phaster.ca/) 30 was used. To identify Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)–Cas systems, CRISPR–Cas Meta Finder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasMeta/Index) was used. 31
Prediction of Bacteriocin and RiPP
BActeriocin GEnome mining tool (BAGEL4) (http://bagel4.molgenrug.nl/) was used to identify and annotate bacteriocins and ribosomally synthesized and posttranslationally modified peptides (RiPPs) in the B. producta genome. The analysis focused on detecting regions encoding these antimicrobial peptides, which are essential for the probiotic properties of B. producta.
PHYLOGENETIC ANALYSIS
Using the Mash algorithm in the PATRIC database, we identified genes with high sequence similarity to our target gene based on 16S rRNA gene data curated via the National Center for Biotechnology Information. The 16S rRNA gene was aligned with that of the target genome of selected close relatives using MAFFT software (https://www.ebi.ac.uk/jdispatcher/msa/mafft). 32,33 Finally, FASTTREE (v2.1) and RaxML software (v8) were used for phylogenetic tree inference via the maximum likelihood method to statistically estimate evolutionary relationships among the taxa. 34
Results
OVERALL GENOME AND COMPARATIVE GENOME ASSEMBLY
The B. producta genome assembly was reported previously to be 100% complete with 3.8% contamination, suggesting a relatively clean assembly. The overall assembly had 120 contigs, collectively spanning 5,965,788 bp, with an average of 45.91% G+C content. The highest N50 metric was detected for the 142,259 bp nestles, which typically signifies good contiguity and assembly quality (Table 1). 35 The RASTtk predicted a total of 215 subsystems, which included various functional and RNA genes (Fig. 1). For comparative studies, the bandage plots showed disparities, but Unicycler had superior assembly characteristics (Table 2). In contrast, BUSCO did not signify an optimal assembler choice due to the striking distinction between the assemblies. The quality analysis by QUAST favors Unicycler over SPAdes in key metrics such as N50, L50, L90, and auN, which is attributed to its ability to generate longer contigs and achieve greater contiguity. 36 However, SPAdes exhibited superior performance in terms of metrics such as the largest contig, total length, and N90.
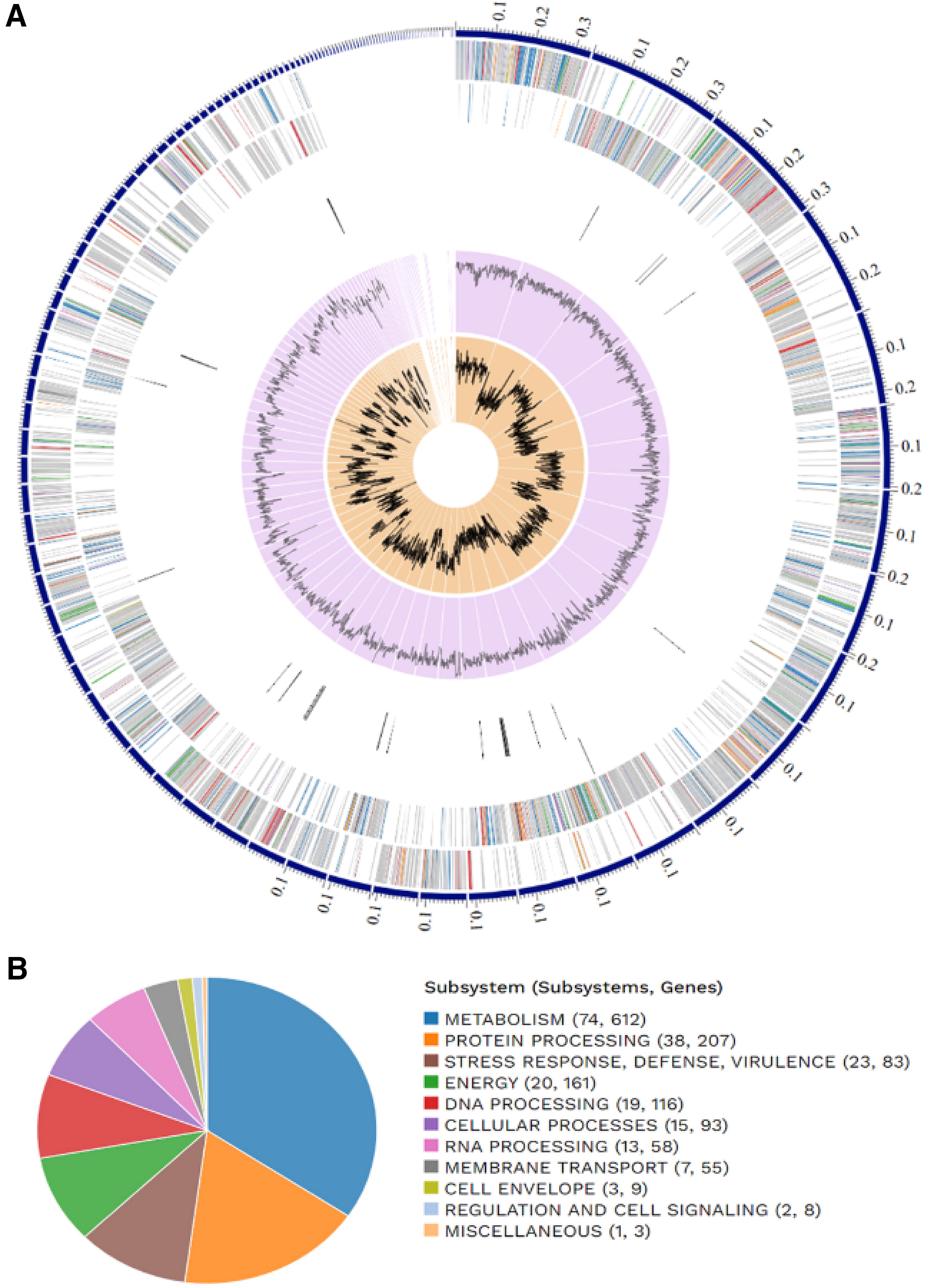
General Blautia producta Genomic Features
Comparison of Bandage Plots: Unicycler vs. SPAdes Assembly.
FUNCTIONAL ANNOTATION AND GENOMIC CHARACTERIZATION
OGs of protein and CAZymes
In total, 4630 proteins were assigned to 64 COG categories by the EggNOG mapper, and the 15 most abundant categories were selected (Fig. 2A; Table 3). The distribution highlights the diverse functionalities encoded within the B. producta genome. The presence of categories such as carbohydrate transport and metabolism, energy production, and signal transduction mechanisms aligns with B. producta’s potential probiotic properties. In KEGG, most entries with an e-value of 0.0 indicate high confidence in the functional annotations (Fig. 3). Additionally, analysis of the score distribution indicated that the majority of the sequence alignments had scores between 0 and 1000. This suggests a moderate-to-high level of similarity between B. producta sequences and database entries, further supporting the reliability of the sequence matches. Figure 2B annotated B. producta proteins identified in a range of metabolic pathways relevant to its probiotic properties (Table 4). Notably, pathways involved in nutrient transport (ko02010—ABC transporters), protein synthesis (ko03010—ribosome), and energy production (ko00190—oxidative phosphorylation) were enriched (Supplementary Figure S1). In addition, pathways for essential building block synthesis, such as aminoacyl-tRNA biosynthesis (ko00970) and biosynthesis of amino acids (ko01230) (Supplementary Figures S2, S3, and S4), were identified, indicating the self-sufficiency of B. producta. Furthermore, the presence of pathways for pentose and glucuronate interconversions (ko00040) suggested that B. producta can utilize various carbohydrate sources (Supplementary Figure S5). Interestingly, pathways related to quorum sensing (ko02024) and the biosynthesis of secondary metabolites (ko01110) were also found (Supplementary Figure S6). These pathways might play a role in B. producta communication with other gut bacteria and potentially contribute to its overall impact on the gut environment.


Distribution of e-values and scores. The e-value distribution provides insight into the statistical significance of the protein alignments, with lower values indicating more significant matches. The score distribution shows the quality of alignment, with higher scores reflecting stronger similarity to known sequences.
Functions of 15 Most Frequently Occurring Clusters of Orthologous Groups
COG, Clusters of Orthologous Groups.
Top 10 Most Frequent Occurring KEGG Pathway
KEGG, Kyoto Encyclopedia of Genes and Genomes.
In contrast, there are diverse CAZyme categories among the annotated proteins that were identified. The most abundant glycoside hydrolase (GH) enzymes were GH3, GH31, and GH13. In addition, GH5, GH43, and GH94 were associated with the various polysaccharide’s digestion (PMID: 25533455 for GH3, PMID: 22992189 for GH5, PMID: 17085431 for GH13, PMID: 36806678 for GH31, PMID: 26729713 for GH43, and PMID: 15274915 for GH94). Similarly, more prominent structural glycosyltransferase (GT) enzymes were GT4 and GT2, and carbohydrate-binding module (CBM), CBM48, was also identified (Fig. 4), indicating a B. producta adaptation to the gut environment. The remaining identified CAZyme categories among queries are illustrated in Fig. 4.

Distribution of Blautia producta CAZyme categories identified in B. producta CB7BLD4. The categories include glycoside hydrolases (GHs), glycosyl transferases (GTs), and carbohydrate-binding module (CBM).
Secondary metabolites
antiSMASH identified two intriguing secondary metabolite gene clusters in the B. producta genome. Region 1.1 exhibited a nonribosomal peptide synthetase (Nonribosomal Peptide Synthetase NRPS) cluster with low similarity to known clusters, suggesting a novel functional peptide. This encodes D-alanyl-D-alanine carboxypeptidase, a multi-antimicrobial extrusion (MATE) efflux pump, and a major facilitator superfamily (MFS) efflux pump. Both MATE and MFS contribute to the multidrug resistance phenotype by efflux of several antimicrobial compounds from the cells, protection against cell wall damage, and homeostasis regulations. Another region, 10.1, contains a ranthipeptide cluster known for diverse functions, including antimicrobial and signaling roles. The identified ranthipeptide shows moderate similarity to known clusters, suggesting potentially unique functions. 36,37 Interestingly, antiSMASH also revealed that region 17.1 harbors a putative cyclic lactone autoinducer cluster, characterized by the presence of genes encoding a putative biosynthase (TIGR04223) and a potential efflux ABC transporter. In addition, the presence of an AraC-family transcriptional regulator in cluster involved in important cellular regulatory mechanism. This gene structuring and inferred functions are similar to the well-studied cyclic lactone autoinducer systems of other related bacterial species. 38 Although experimental validation is required to confirm the function of this cluster, an in silico analysis has highlighted its potential involvement in quorum sensing (Fig. 5). 39,40
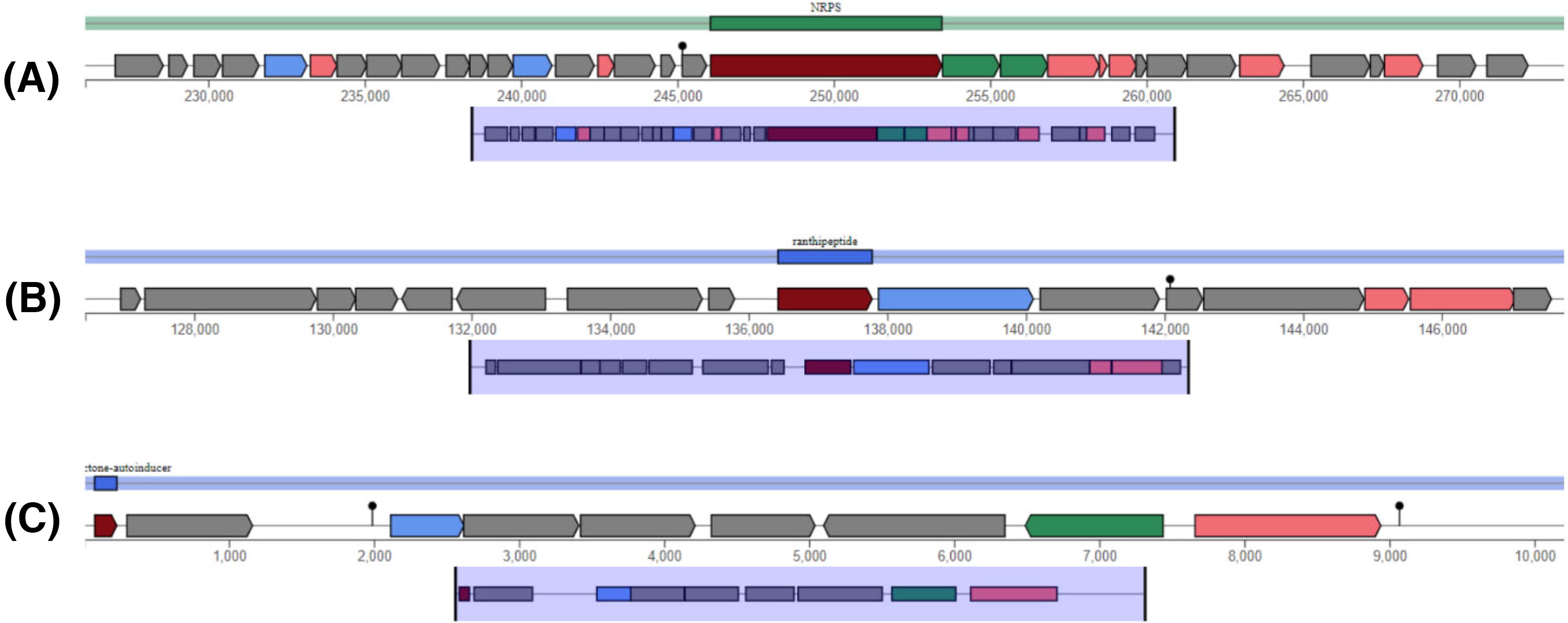
AntiSMASH regions with gene clusters for secondary metabolites in Blautia producta CB7BLD4. The arrows indicate the direction of transcription for each gene, and the colors indicate gene function.
Resistance and prophage genes
The antimicrobial resistance (AMR) of B. producta was studied using ResFinder, and the results indicated that it was not resistant to any of the 116 antimicrobial agents, suggesting its probiotic candidacy. Furthermore, due to the non-AMR genes, there is minimal risk of horizontal gene transfer and antibiotic resistance dissemination within the gut microbiome. These findings further support the safety profile of the B. producta strain and its suitability for probiotic applications. PathogenFinder (Fig. 6) suggested that B. producta has low human pathogenicity (42.7%), while three out of seven sequences (in red) matched known human pathogens; these sequences represented less closely related taxa. Nevertheless, four sequences (in green) showed high identity (86.91% average) to known gut commensals, including Clostridium saccharolyticum (a butyrate producer), C. cellulolyticum (a cellulose degrader), and C. phytofermentans (a lactic acid producer). However, three matches (in red) need further investigation into specific virulence factors and safety assessments before being considered probiotic.
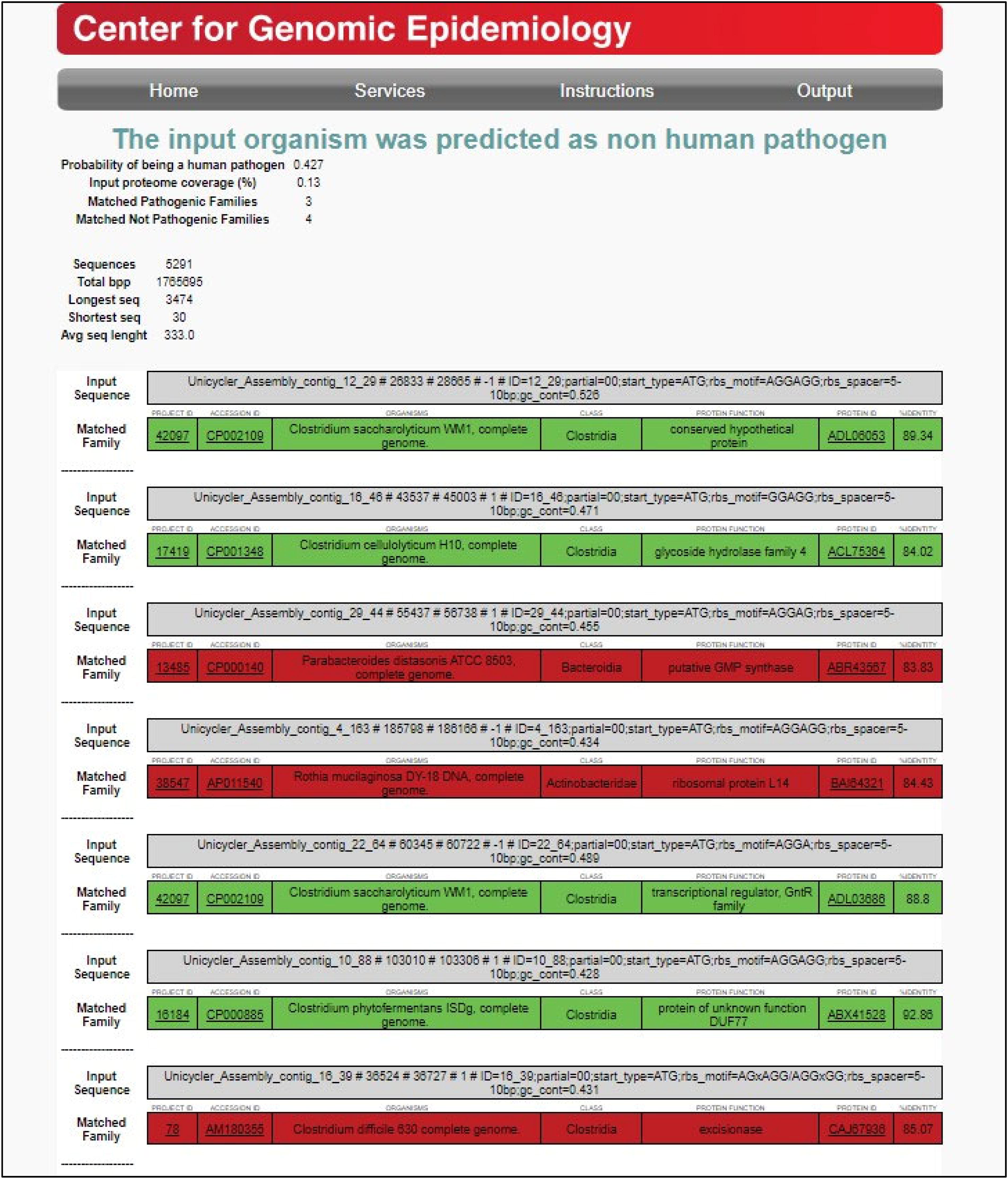
PathogenFinder for pathogen matches for Blautia producta CB7BLD4. Results indicate a low human pathogenicity score of 42.7%, suggesting minimal risk of pathogenicity.
Furthermore, CARD has shown no perfect hits for known antibiotic resistance genes (ARGs). Two strict hits associated with glycopeptide resistance were identified for the putative vanT and vanY genes (Fig. 7). These genes share sequence similarity to known resistance genes. The reciprocal BLAST analysis revealed that they are not strict orthologs, and these findings suggested that their potential role in glycopeptide resistance may be limited or that they might have evolved independently. Further experimental validation is required to confirm their functional significance. Although these hits met the CARD criteria for a potential resistance function, their low percentage identity (35–39%) and nonorthologous relationship suggest possible nonfunctional remnants or unrelated roles. These ambiguities require further assessment of glycopeptide resistance in B. producta. Moreover, PHASTER identified functional prophages in the region two with similar characteristics to those of an intact phage. This finding influences B. producta adaptability, virulence, and interactions with other organisms. While the presence of prophages can be advantageous for bacteria through enhanced adaptation, horizontal gene transfer, and even direct contributions to probiotic features such as bacteriocin production, potential drawbacks exist. These include virulence acquisition through phage gene transfer, reduced fitness due to metabolic burdens, and unpredictable interactions with environmental factors that could trigger prophage induction and alter B. producta behavior. Further investigation of the specific genes encoded by the prophage, its induction conditions, and its performance in simulated and real-world gut environments will be crucial to determine its net impact on B. producta probiotic properties (Fig. 8).
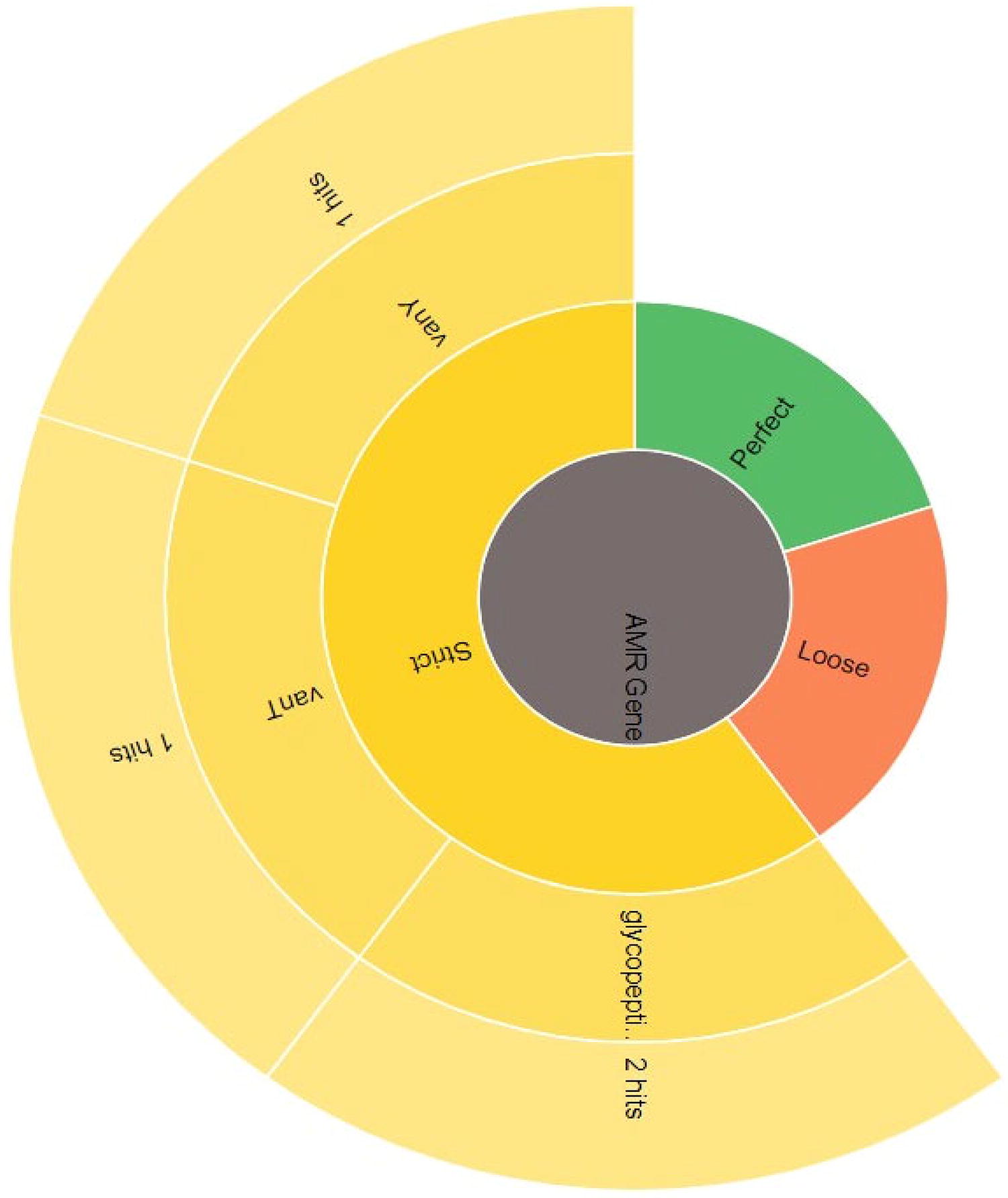
CARD results showing the AMR gene family. Two strict hits, potentially associated with glycopeptide resistance, were identified for the vanT and vanY genes, although not strict orthologs. Their low identity percentages (35–39%) to known resistance genes suggest these may be nonfunctional remnants or genes with unrelated roles. AMR, antimicrobial resistance; CARD, Comprehensive Antibiotic Resistance Database.
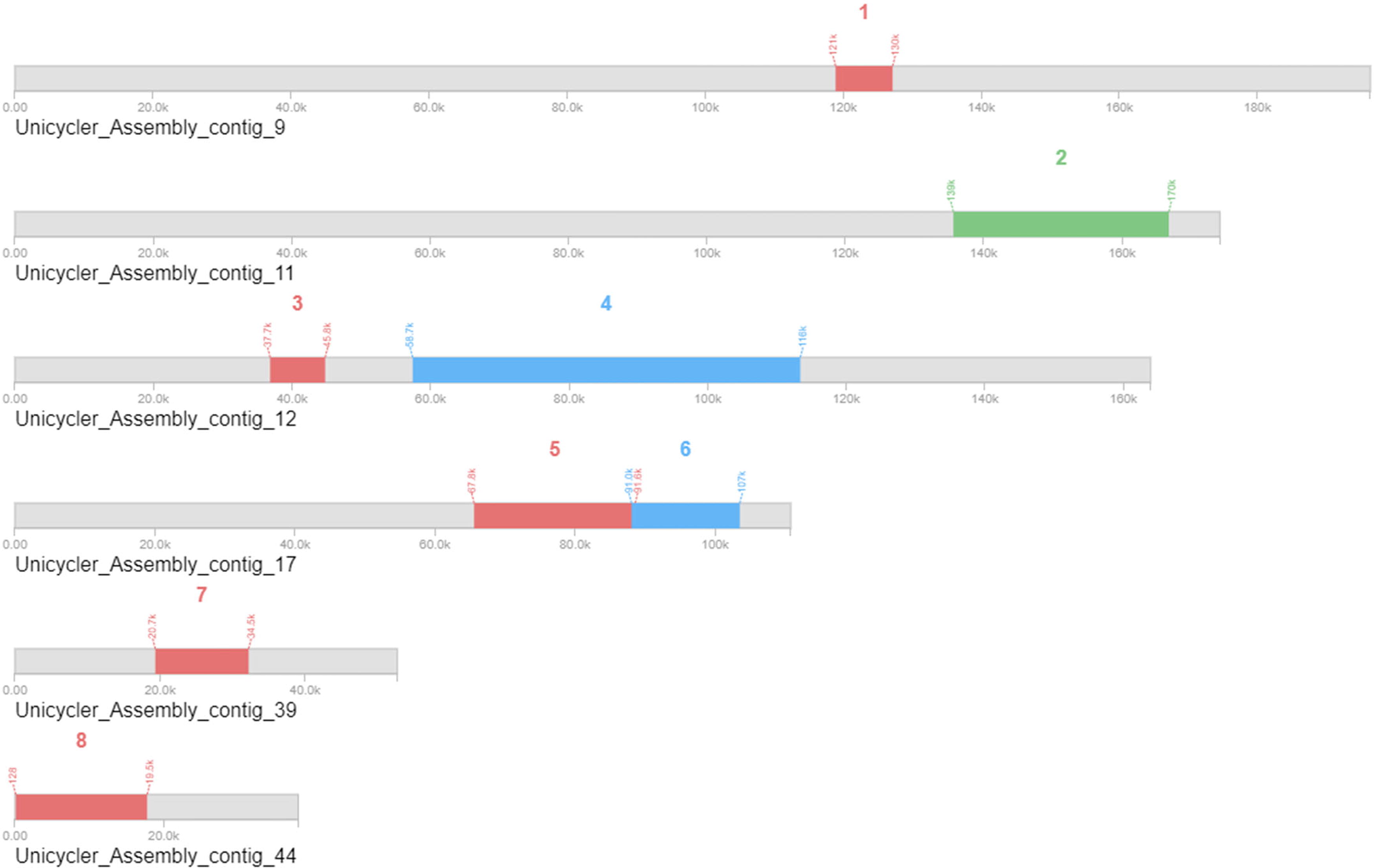
PHASTER results showing eight identified prophage regions, with region 2 showing characteristics similar to intact, functional phages. PHASTER, PHAGE Search Tool Enhanced Release.
CRISPR–Cas systems
CRISPR–Cas systems revealed that 10 out of 22 analyzed sequences contained CRISPR arrays, suggesting the presence of this antiviral defense mechanism. Interestingly, Cas genes, which are crucial for the function of the CRISPR–Cas system, were found in only five sequences. This indicates that some sequences might possess CRISPR arrays but rely on Cas genes from elsewhere in the genome, whereas others have Cas genes but lack CRISPR arrays, potentially due to recent acquisition or loss or incompleteness of genomic data for our organism. These findings highlight the diversity of the CRISPR–Cas system distribution within the analyzed sequences of B. producta, suggesting that the strains were probiotic positive (Fig. 9).

CRISPR—Cas Meta results comprising 10 CRISPR arrays and 5 Cas systems across 22 sequences in the Blautia producta CB7BLD4 genome.
Prediction of Bacteriocin and RiPP
The two areas of interest containing genes for biosynthesis of sactipeptides, which are a subclass of RiPPs with antimicrobial potential, were identified by BAGEL4. Many biosynthetic genes within these loci may be involved in bacteriocin production and transport, including genes encoding ABC transporters and several hypothetical proteins. BmbF (subtilosin biosynthesis protein) is a prominent protein in bacteriocin pathways (Fig. 10), which implies potential for B. producta bacteriocins production. Furthermore, the bacteriocins production and antimicrobial activity must be validated within the laboratory conditions.

Representation of gene clusters coding the production of two sactipeptides
PHYLOGENETIC ANALYSIS
Figure 11 shows that B. producta had close similarities with B. producta ATCC 27340, with a bootstrap support value of 100. The next closest relative was Blautia sp. KLE 1732, with a bootstrap support value of 42.

Phylogenetic tree of Blautia producta CB7BLD4 shows a close relationship between B. producta CB7BLD4 and B. producta ATCC 27340, supported by a bootstrap value of 100, indicating high confidence. The next closest relative identified was Blautia sp. KLE 1732, with a lower bootstrap value of 42, suggesting less certainty in its placement within the tree.
Discussion
Comparative results of different probiotic strains within B. producta along with other well-known probiotic species provided in Supplementary Figure S1A and S1B support its probiotic candidacy. Similar subsystems, such as metabolism, stress response, and energy production, were also identified in other probiotic strains. Unlike B. producta, Lactococcus lactis has 246 subsystems related to protein and carbohydrate metabolism, vitamin biosynthesis, and amino acid metabolism, showing its survival and beneficial effects in the gut. 41 Other prominent GIT probiotic members, such as Bifidobacterium longum and Enterococcus faecalis, have OGs similar to those of B. producta, suggesting their roles in carbohydrate transport, metabolism, signal transduction, and defense. 42,43 The pathways for nutrient transport and amino acid biosynthesis in B. producta revealed that metabolic processes involving dietary sugars are similar to those in Streptococcus thermophilus. 44 B. producta CAZymes, in contrast, for GH and GT are also found in other probiotics, such as Pediococcus pentosaceus and Bacillus subtilis, influencing the gut microbiota composition. 45,46
Furthermore, the secondary metabolite gene clusters in B. producta, including a putative NRPS cluster with antimicrobial functions, were found to be similar to the bacteriocinogenic clusters of L. lactis, suggesting that both have antimicrobial compounds for gut health. 41 Similar to other probiotics such as L. lactis and S. thermophilus, the absence of detectable ARGs and plasmids in B. producta can be interpreted for safe probiotic potential compared with strains with ARGs. 41,44 However, even with potentially active prophages, B. producta lacks associated resistance genes, which is similar to the situation in E. faecalis. 43 The presence of CRISPR arrays in B. producta, which defenses against viruses, was found in other probiotics, such as S. thermophilus. However, further confirmation of functional Cas genes has yet to be achieved. 44 The detection of tRNA and rRNA genes in B. producta reflects that its ability to synthesize protein is almost identical to that of B. longum, which emphasizes the credibility of the sequencing data. 42
In conclusion, the role of B. producta as a probiotic candidate was assessed through WGS. The high-quality assembly identified various functional categories within its genome, including those related to carbohydrate transport and metabolism, energy, and signal transduction, which are crucial for any probiotic function in the gut. KEGG analysis revealed pathways involved in nutrient transport, protein synthesis, and energy production, which are necessary for gut adaptability. Intriguingly, the gene clusters for quorum sensing and secondary metabolites were also found to be beneficial to the overall gut ecosystem. The absence of resistance genes in B. producta also minimizes the risk of human pathogenicity, suggesting that this strain is highly safe. However, the presence of potentially functional prophages without functional genes will determine their impact on probiotic properties. In CRISPR–Cas systems, several sequences were identified for antiviral defense, which validated its probiotic potential. In addition to our analysis, further research is needed on the specific mechanisms underlying its probiotic effects and both in vivo and computational evaluation of B. producta in animal models for various health conditions.
Footnotes
Acknowledgments
All the authors acknowledge the support of Dr. Ashok Chouhan, Found President, Amity University Uttar Pradesh, Noida, India, for his encouragement and support. The authors would also like to thank Prof. Dr. Pooja Vijayaraghavan and Dr. Shivayogeeswar Neelagund for enduring support throughout the research.
Authors’ Contributions
A.T.: Experimental design, data collection and interpretation, and draft writing. A.N.: Data collection and interpretation and draft writing. D.P.: Critical evaluation of data and interpretation. K.K.P.: Interpretation and draft writing. R.K.S.: Lab resources and draft writing. G.D.M.: Supervision, experimental design, evaluation and interpretation, laboratory resources, and manuscript revision. P.K.: Supervision, laboratory resources, and manuscript revision.
Data availability
The article contains all the data generated during this study.
Ethical Approval
No ethical approval was needed, as no live animals were used in this study.
Author Disclosure Statement
No competing interests exist.
Funding Information
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.