
Abstract
This article presents a “green” approach to water-soluble oligohistidine peptides through protease-catalyzed peptide synthesis. Synthesis was catalyzed by papain and α-chymotrypsin, and reactions were monitored in-situ by real-time 1-hour NMR spectroscopy. Pyrosetta computational modeling was used to calculate the energy required for amidation versus hydrolysis with papain and α-chymotrypsin.
Histidine is an essential amino acid that provides unique chemical properties. Its imidazole ring is amphoteric, enabling it to function within the physiological pH range; it is soluble at lower pH (∼6.3) and insoluble at physiological pH (7.4). 1 The significance of its aromatic ring system is reflected in its wide occurrence in natural compounds such as purines, histamine, and various classes of pharmaceuticals. 2 Imidazole containing compounds can provide anti-inflammatory, antioxidant, anti-secretory, metal remediation, and therapeutic properties. 1,3 –5 Despite the many valuable applications of histidine-rich peptides, synthetic options remain limited. Histidine-containing peptides are most frequently prepared by solid-phase peptide synthesis, ring-opening polymerization of N-carboxyanhydrides, and LPPS. 4 A green approach to histidine-rich peptides has yet to be developed.
Recently, Terada et al. (2022) reported the protease-catalyzed peptide synthesis (PCPS) of His-containing co-oligopeptides using papain as a catalyst. 6 Papain-catalyzed oligomerization of His-OEt was reported to not be successful based on the absence of precipitate formation. Apparently, Terada et al. did not analyze potential formation of oligo(His) in the aqueous phase. This article revisits the work of Terada et al. 6 on crude papain-catalyzed oligomerization of His-OEt. Parallel studies on His-OEt oligomerization were also carried out using α-chymotrypsin as a catalyst. Reactions were monitored in real-time by in-situ NMR, and product chain lengths were analyzed by Electrospray Ionization Mass Spectrometry (ESI-MS). Computational modeling was performed through PyRosetta software to rationalize the observed differences in oligo(His) formation by crude papain and α-chymotrypsin. This work demonstrates a lower-cost approach to prepare water-soluble oligo(His) and provides information on protease structure-activity relationships for His-OEt oligomerization by PCPS.
Papain is a cysteine protease isolated from the papaya plant. Given the promiscuous ability of papain to catalyze oligomerizations of amino acid ester monomers, 7 we aim to revisit papain-catalyzed His-OEt. In addition, α-chymotrypsin, a constituent of pancreatic juice, has also proved active for numerous amino acid ester monomer oligomerizations, including L-Cys-OEt, L-Glu-(OEt)2, L-Tyr-OMe, D-amino acid methyl esters (Met, Ser, Phe, Arg, Val, Leu, Ala), L-Phe-OEt, L-Leu-OEt, L-Met-OEt, L-Trp-OEt, and Lys-Leu-OEt. 8 –13
Structures of His-OEt monomer, free acid His-OH, and oligo(His)x are depicted in Figure 1 with letter designations for correlation with NMR signals, shown in Figure 2–3, Supplementary Figure S2–S3. 1H NMR spectral peaks were identified based on COSY NMR analysis. In addition, by overlaying the 1H NMR spectrum of free acid His-OH with 1H NMR spectra of His-OEt oligomerization reactions, free acid monomer peaks were identified (Supplementary Fig. S2 and S3).
Monomer conversion, or the percent of monomer converted to peptide repeat units in a predetermined time, was determined by 1H NMR, displayed in Figure 2–3. Conversion was calculated by dividing the oligopeptide observed by NMR by the sum of oligopeptide plus free acid (His-OH) present. This calculates how much of the starting material was converted to oligopeptide versus hydrolyzed into free acid, His-OH. By 1H NMR, only the monomer in its free acid form, His-OH (signal dʺ), is present after the start of the reaction, indicating there is no unreacted His-OEt monomer left in the reactions, and it was either converted to oligopeptide or hydrolyzed to free acid (SI, eq. 1).
1H NMR spectroscopy was used to monitor oligo(His) peptide formation, catalyzed by papain and α-chymotrypsin, as a function of time (Fig. 2, 3). Values of %-monomer conversion at <10 minutes could not be obtained due to the time required to lock and shim samples on the NMR instrument. As can be observed in Supplementary Figure S4, the progression of the time course of %-monomer conversion versus time for papain- and α-chymotrypsin-catalyzed His-OEt oligomerizations substantially differ. For papain, in ≤10 minutes, the %conversion of monomer to peptide reaches 55 ± 7.4%. From 10 to 60 minutes, the %-monomer conversion did not significantly change.
For α-chymotrypsin, in ≤10 minutes, the %-conversion of monomer to oligo(His) reaches 69 ± 9.9% within 10 minutes. However, from 10 to 60 minutes, peptide hydrolysis occurs such that monomer concentration in reaction solutions increases and %-monomer conversion to oligo(His) decreases to 14 ± 7.4%. This suggests that the rate of amidation to hydrolysis for His-OEt oligomerizations is higher for papain relative to α-chymotrypsin catalysis. In both papain- and α-chymotrypsin-catalyzed His-OEt oligomerizations, the products formed remained soluble in the reaction media during the 60-minutes oligomerization. The large standard deviation values denoted by error bars in Supplementary Figure S4 are consistent with variations in the time needed during NMR analysis to lock solvents and shim samples. This led to a difference of ±30 seconds for the start time of each recorded spectrum. Nevertheless, identical trends were observed for all three replicates within each enzyme study.
ESI-MS analysis was used to determine oligo(His) chain lengths present in the reaction as a function of incubation time. High-performance liquid chromatography was attempted but was unsuccessful. This is likely due to the high metal chelation of His units along oligo(His) oligomers that results in its adherence to LC columns. Electrospray ionization (ESI) is a technique that produces ions by application of a high voltage to a liquid-generating an aerosol. Since this method generally overcomes the tendency of biomolecules to fragment, we used ESI in combination with mass spectrometry (MS) to analyze the chain lengths (average degree of polymerization, DPavg) of oligo(His).
Except for the time point at 45 minutes, DPavg values for oligo(His) synthesized by papain catalysis were higher than those resulting from α-chymotrypsin catalysis (Table 1, Supplementary Fig. S5). For papain-catalyzed reactions at 5, 10, and 15 minutes, irregular fluctuations of DPavg (7.2, 4.5, and 9.3, respectively) were observed with unexpectedly narrow standard deviations (Table 1, Supplementary Fig. S5). As the time of papain-catalyzed oligomerization reactions progressed from 15 to 45 minutes, DPavg values decreased from 9.3 to 2.2. This is likely due to papain-catalyzed chain cleavage (i.e., hydrolysis) that corresponds to the low availability of His-OEt by 10 minutes (Fig. 3) such that chain hydrolysis dominates chain growth reactions. Furthermore, since the %-monomer conversion in the reaction media did not change from 10 minutes to 60 minutes for papain-catalyzed oligo(His) synthesis, this indicates that decreases in DPavg, from 15 to 45 minutes, occur by intrachain cleavage events as opposed to exo-cleavage that would generate His-OH.
DPavg Of Oligo(His) at Various Time Increments During Peptide Synthesis for Reactions Catalyzed with (a) Papain and (b) α-Chymotrypsin
In contrast to papain, α-chymotrypsin yielded oligo(His) with consistently short DPavg values that ranged from 2.4 ± 0.08 to 3.4 ± 0.19 as the reaction progressed from 5 to 60 minutes. Also, using α-chymotrypsin resulted in a decrease in %-monomer conversion to oligo(His) from 69 ± 9.9% to 14 ± 7.4% from 10 to 60 minutes, respectively. Thus, simultaneous events of α-chymotrypsin chain cleavage and chain formation must occur such that oligo(His) chain lengths remain at DPavg 3.
To investigate the competition between peptide synthesis and hydrolysis for papain and α-chymotrypsin, computational modeling was performed with Rosetta. Acyl-enzyme intermediates were modeled with a His acyl-donor and His acyl-acceptor pair to represent synthesis and a His-His dipeptide to represent hydrolysis (Fig. 4). The substrates are represented as single monomers or dipeptides in the models but may exist in solution as short oligopeptides.
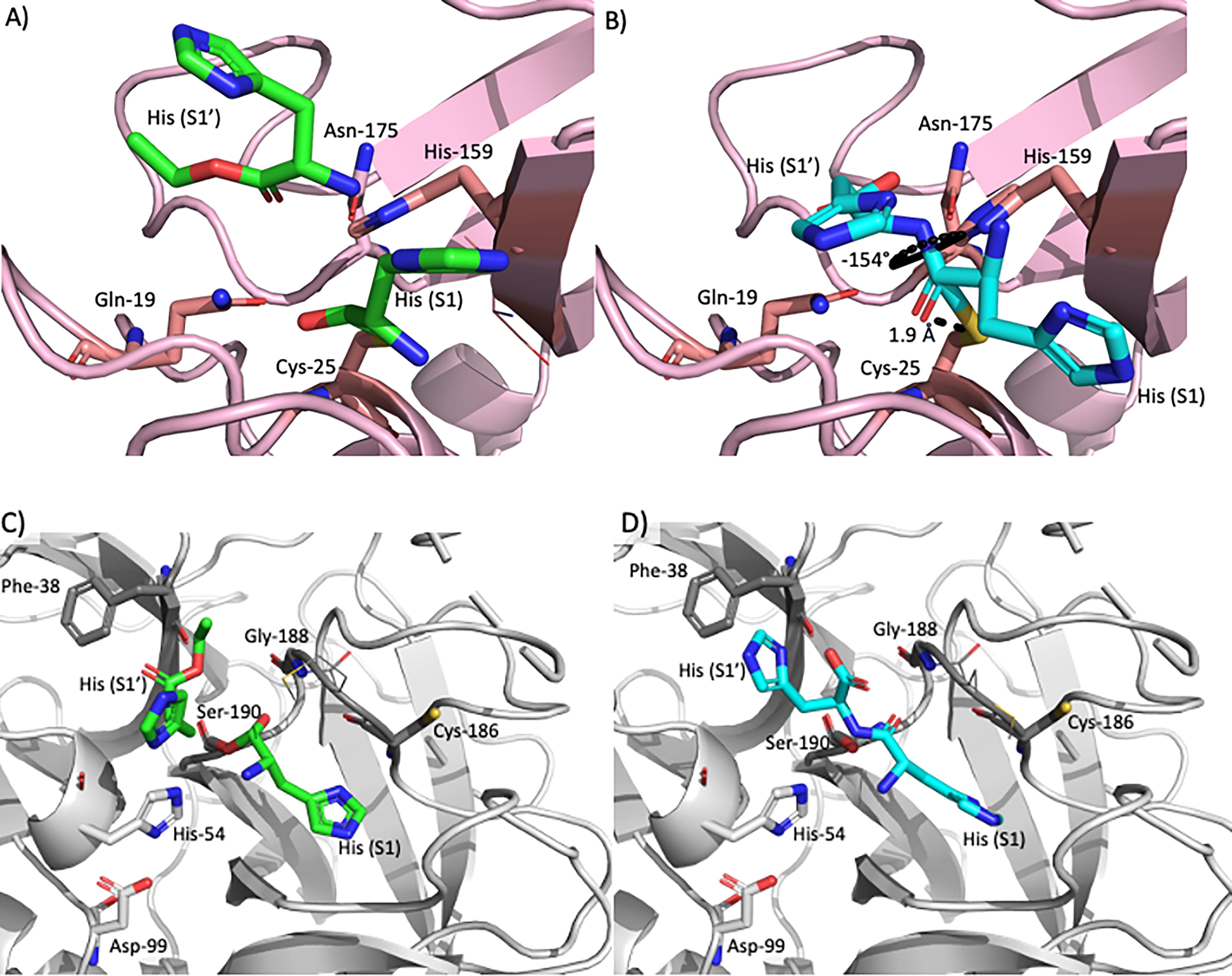
Active site docking models for papain (synthesis in
Energy scores of the active site shell residues in various states were used as proxies for activity. These shell energies were computed based on interactions between residues in the acyl-enzyme intermediate state. The transition state geometries were maintained by constraining the positions of the substrates relative to the active site. Energies were considered for the entire pocket, which includes the substrates, and for the pocket residues only, which does not include the substrates.
For papain, the energies are more favorable with the monomer substrates for synthesis than with the dipeptide substrate for hydrolysis (Supplementary Fig. S6). This difference agrees with the preference for synthesis observed with papain under conditions (at early time points) when sufficient substrates are available to serve as acyl acceptors and acyl donors. However, hydrolysis is not entirely unfavorable, suggesting that with time some of the synthesized oligopeptides (and potentially His-OEt monomers) are expected to be hydrolyzed. Hydrolysis over time leads to an increase in the concentration of EtOH (Fig. 2) and a decrease in the concentration of acyl donor His-OEt entities; therefore, the DP values are expected to decrease with time due to hydrolysis. As synthesis is expected to be significantly energetically favorable compared to hydrolysis and the pool of available His-OEt is large, the generated nucleophilic acyl acceptors due to hydrolysis are poised to react with His-OEt monomers and oligomers and lead to an increase in DP over time. The balance between these competing effects leads to oscillations in DPavg (Supplementary Fig. S5).
For α-chymotrypsin, the substrate-bound energies for both hydrolysis and bond formation are comparable, whereas the active site residues are more stabilized in the presence of the dipeptide than the monomers (Supplementary Fig. S6), suggesting that hydrolysis is slightly favored over synthesis. The similarly favorable energies for both synthesis and hydrolysis do not lead to dominance of one over the other with time, as observed with papain, and agree with the observed trend of simultaneous events of α-chymotrypsin chain cleavage and chain formation that occur such that oligo(His) chain lengths remain at DPavg 3 (Supplementary Fig. S5).
Individual residues within the active sites were compared to further investigate the origin of these energy differences (Supplementary Fig. S6). In papain, the substrates Gln-19, Cys-25, and His-159 were found to be more destabilized when docked with a dipeptide (hydrolysis) than with the monomers (synthesis). As for α-chymotrypsin, Ser-190, Cys-186, Phe-38, and Gly-188 were slightly more destabilized in the presence of monomers (synthesis) compared to the dipeptide (hydrolysis; Supplementary Fig. S6).
For papain, the moiety with the largest energy difference was the His in the S1 binding site, which would be either the acyl donor in the synthesis model or the first His in the dipeptide in the hydrolysis model (Supplementary Fig. S6). Breaking down the scores into individual score terms reveals that the largest difference is due to the distortion of the dipeptide, namely the omega bond between the two His substrates. This indicates that the His-His dipeptide adopts a slightly unfavorable omega bond and thus conformation to fit properly in the papain active site for hydrolysis. Additionally, the active site residues have more favorable energies with the monomers (synthesis) than with the dipeptide (hydrolysis). The positioning of the dipeptide places the S1 His backbone carbonyl oxygen near the sulfur on catalytic Cys-25, thus introducing repulsion that is not present with the monomers.
For α-chymotrypsin, the monomers are more stabilized than dipeptides, but active site residues are more stabilized with dipeptides than with the monomers (Supplementary Fig. S6). The residues with the largest energy differences between the models are Ser-190, Cys-186, Phe-38, and Gly-188, two of which are catalytic residues. All four are more destabilized with the monomers than with the peptide, which is contributed by higher repulsion with the monomers. For instance, the distances between the sidechain and ester of the S1′ His and the sidechain and backbone of Phe-38 are more favorable than from the His acyl-acceptor, and the rotamer of the sidechain on the S1 His in the dipeptide is more favorable for Cys-186 than the rotamer for the His acyl-donor.
These results provide insight into the molecular origin of the observed experimental preferences of the two enzymes for hydrolysis and synthesis.
In conclusion, water-soluble histidine oligopeptides were successfully synthesized in situ by protease-catalyzed peptide synthesis, using papain and α-chymotrypsin as catalyst. In summary, papain yielded 55 ± 7.4% monomer conversion within 10 minutes and remained stable. α-Chymotrypsin, however, produced a peptide yield of 69 ± 9.9% within 10 minutes and then proceeded to hydrolyze the peptides. Monomer conversion to peptide was measured by 1H NMR spectroscopy, and the average degree of polymerization (DPavg) of oligo(His) was determined by ESI spectrometry. According to DPavg analysis, oligohistidine underwent an extent of hydrolysis with both papain and α-chymotrypsin as catalysts, but papain yielded oligohistidine chains with much higher DPavg than that of α-chymotrypsin, with maximum chain lengths of 9.3 ± 0.05 and 3.4 ± 0.19, respectively. Finally, computational modeling conducted by Rosetta software supports the experimental results by demonstrating that when papain is catalysts, amidation is more energetically favorable than that of α-chymotrypsin.
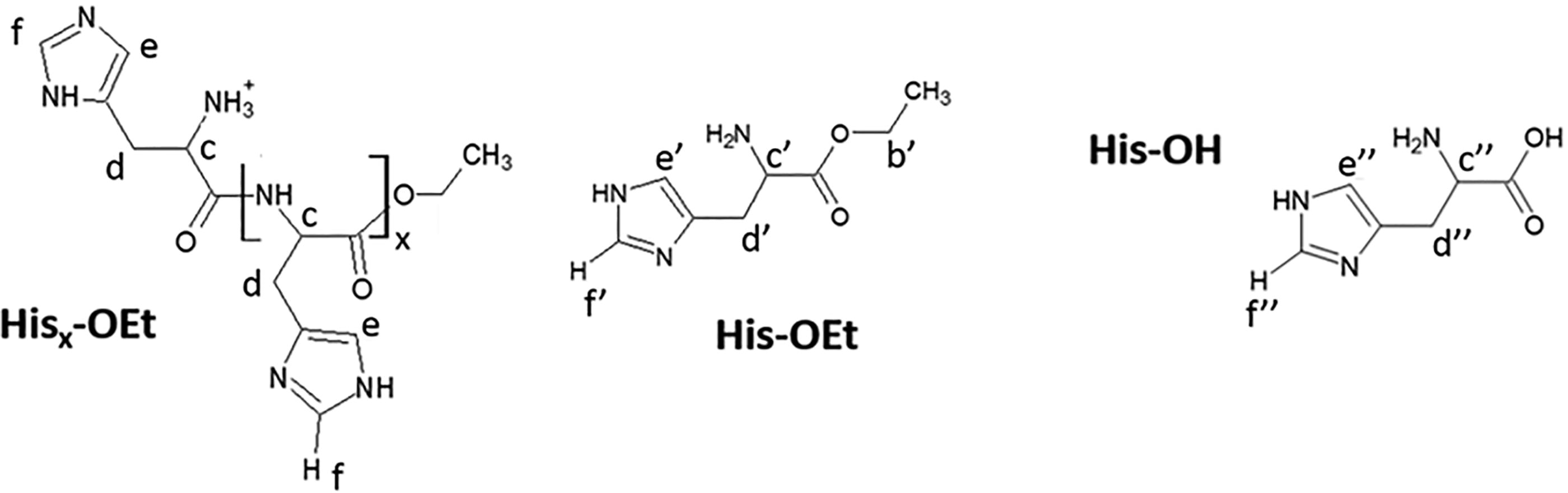
Structures of oligo(His), His-OEt monomer, and free acid His-OH with letter designations for correlation with NMR signals displayed in Figs. 2–3, S2–S3.
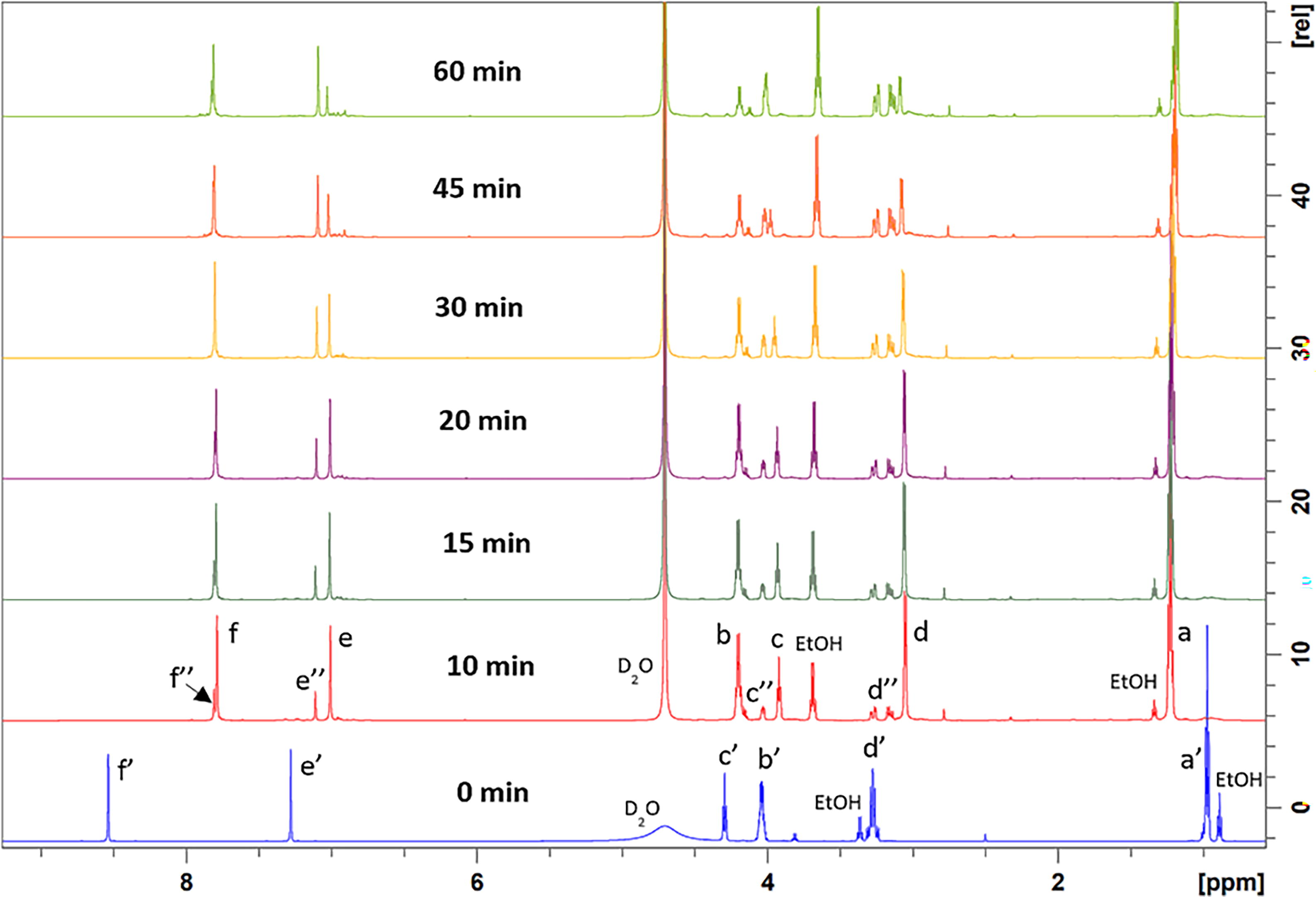
1H NMR spectra at (600 MHz, D2O) recorded at times up to 60 minutes to monitor the progression of papain-catalyzed His-OEt oligomerization.

1H NMR spectra (600 MHz, D2O) recorded at times up to 60 minutes to monitor the progression of α-chymotrypsin-catalyzed His-OEt oligomerization.
Footnotes
Authors’ Contributions
S.P.B.: Data curation, formal analysis, original draft writing. M.L.: Data curation, software, original draft writing. K.X.: Data curation. S.D.K.: Software, supervision, review and editing. R.A.G.: Conceptualization, supervision, review and editing.
Data Availability
The data supporting this article has been included as part of the Supplementary Information.
Author Disclosure Statement
Parts of this article are based on research previously presented in the dissertation, “Protease-catalyzed synthesis of oligopeptides and glycan-terminated peptides” by Sarah Black, Rensselaer Polytechnic Institute, 2023.
Funding Information
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors thank their funding from
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.