
Abstract
Abstract
Background:
BAY41-6551, a drug–device combination in development for adjunctive treatment of Gram-negative pneumonia in intubated and mechanically ventilated patients, consists of amikacin formulated for inhalation coupled with the Pulmonary Drug Delivery System (PDDS) Clinical aerosol delivery platform. Given the predominantly renal clearance of aminoglycosides, understanding systemic amikacin exposure and safety during administration of BAY41-6551 to patients with acute renal failure (ARF) is clinically important.
Methods:
Seven mechanically ventilated patients with Gram-negative pneumonia and ARF receiving continuous veno-venous hemodiafiltration (CVVHDF) were treated with multiple administrations of BAY41-6551 400 mg amikacin twice daily using the PDDS Clinical on-ventilator device [in addition to standard intravenous (i.v.) antimicrobial therapy]. CVVHDF parameters were recorded and a PK analysis was performed using serum, urine, and bronchoalveolar lavage fluid samples.
Results:
Maximum serum amikacin concentration [median 1.93 (range: 0.63–3.99) mg/L] and area under the concentration–time curve from zero to 12 h on day 3 [median 19.32 (range 6.32–36.87) mg · h/L] were elevated compared with mechanically ventilated patients with normal renal function; however, serum amikacin trough concentrations were within accepted safety limits. The median amikacin concentration in epithelial lining fluid [887 (range: 406–12,819) mg/L] was similar to that reported previously in mechanically ventilated patients with normal renal function. BAY41-6551 demonstrated acceptable safety and tolerability with most adverse events (AEs) as expected for the patient population. One serious AE of bronchospasm was attributed to the study medication; no reported AEs were related to the PDDS Clinical device.
Conclusions:
CVVHDF appears to provide adequate clearance of systemically absorbed amikacin in mechanically ventilated patients with ARF, suggesting that dose adjustments for BAY41-6551 are probably not necessary for this patient population. Nonetheless, the standard precautionary measures for critically ill patients receiving i.v. amikacin should be followed for patients with ARF who are treated with BAY41-6551.
Introduction
BAY41-6551 (Bayer Schering Pharma/Nektar Therapeutics, San Francisco, CA) is a drug–device combination comprising an amikacin solution formulated for inhalation and a proprietary pulmonary aerosol delivery platform [the Pulmonary Drug Delivery System (PDDS) Clinical]. BAY41-6551 is being investigated for adjunctive treatment of Gram-negative pneumonia in mechanically ventilated patients. The PDDS Clinical employs a high-frequency OnQ® piezoelectric vibrating plate to produce a high proportion of aerosol particles within the respirable range (volume median diameter 3–5 μm).(3) It offers dual functionality, either integrated within a standard mechanical ventilator circuit (on-ventilator configuration) with breath actuation controlling the aerosol generation during a defined portion of the inspiratory cycle or fitted to a hand-held inhalation unit (hand-held configuration) with continuous nebulization for patients no longer requiring ventilatory support.(3) For clarity, hereafter both the specifically formulated amikacin delivered by the PDDS Clinical and the drug–device combination itself are referred to as BAY41-6551.
A potential advantage of inhaled antibiotic administration for treating acute pulmonary infections is the attainment of very high local drug concentrations at infected tissue sites with limited systemic drug exposure; this is particularly desirable for concentration-dependent antibiotics such as aminoglycosides,(4) which may be associated with dose-limiting toxicity when administered by the intravenous (i.v.) route.(5) Aminoglycosides are well suited to inhaled delivery, having a good spectrum of activity against most respiratory tract pathogens, synergy with β-lactam antibiotics (which form a cornerstone of i.v. antibiotic treatment guidelines for nosocomial pneumonia),(2) and acceptable taste.(6) However, despite significant clinical experience with nebulized antibiotics, particularly tobramycin and colistin for patients with cystic fibrosis, there is currently limited evidence on the clinical utility of this mode of administration for critically ill patients.(2)
Phase I and II studies in healthy volunteers(7,8) and mechanically ventilated patients with Gram-negative pneumonia(9,10) but without renal impairment (i.e., with creatinine clearance ≥80 mL/min) have shown that BAY41-6551 is well tolerated at doses up to 400 mg amikacin twice daily (b.i.d.). A previous study in mechanically ventilated patients showed that 400 mg b.i.d. BAY41-6551 achieved tracheal aspirate amikacin concentrations 5000 times higher than those in serum and over 1000 times higher than historical data for amikacin concentrations in bronchial secretions following i.v. amikacin (15 mg/kg/day).(8,11) However, the predominantly renal clearance of aminoglycosides suggests a potential for elevated systemic amikacin exposure following administration of BAY41-6551 to patients with chronic renal insufficiency or acute renal failure (ARF).(5) Given that amikacin in BAY41-6551 is administered to the lungs, where drug absorption is rapid and efficient, and that renal impairment is prevalent in critically ill patients,(12) the impact of ARF and associated renal replacement therapies is an important consideration for BAY41-6551 therapy. In this phase II study, mechanically ventilated patients with Gram-negative pneumonia and ARF treated by continuous veno-venous hemodiafiltration (CVVHDF) received multiple b.i.d. doses of BAY41-6551 containing 400 mg amikacin to assess the impact of ARF on systemic amikacin pharmacokinetics (PK), epithelial lining fluid (ELF) concentrations, and BAY41-6551 safety and tolerability.
Materials and Methods
Study design
This open-label, single-arm PK study, conducted at Hôpital Pitié-Salpêtrière, Paris, France, consisted of a 1- to 2-day screening period, a 7- to 14-day treatment period, and a follow-up visit 25–31 days after the first dose of study medication. The study was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. It was approved by an Independent Ethics Committee at the participating study site; written informed consent was obtained from all participants or their next of kin.
Patients
Adult male and female intubated and mechanically ventilated patients with ARF maintained on CVVHDF who were receiving i.v. antibiotic therapy for nosocomial pneumonia caused by Gram-negative organisms were eligible for enrolment. ARF was defined as oliguria (urine output <200 mL/12 h) despite fluid resuscitation and i.v. diuretic treatment, and/or azotemia [blood urea nitrogen (BUN) >30 mmol/L with urine output <1500 mL/12 h]. Gram-negative pneumonia was defined as the presence of new or progressive infiltrate(s) on chest X-ray and presence of Gram-negative organisms by either culture or Gram stain of respiratory secretions, plus two or more of the following: fever (defined as oral temperature >38.0°C or rectal/core body temperature >39.0°C) or hypothermia (rectal/core body temperature <35.0°C); leukopenia; purulent sputum or respiratory secretions; or a change in the character of sputum. For all patients, the diagnosis of pneumonia was confirmed bacteriologically using quantitative cultures (significant growth defined as ≥104 cfu/mL) of distal bronchoalveolar lavage (BAL)-fluid samples obtained by fiberoptic bronchoscopy.
Patients could be enrolled if they were expected to remain intubated and mechanically ventilated and require treatment for ARF for ≥3 days after the start of study treatment. Reasons for study exclusion included: pregnancy; known local or systemic hypersensitivity to amikacin or aminoglycosides; known respiratory colonization with amikacin-resistant Gram-negative rods; amikacin administration by any route within the previous 7 days; severe hearing loss; current immunosuppressive therapy; severe hypoxemia (defined as alveolar partial pressure of oxygen/fraction of inspired oxygen <200); chronic liver disease; body mass index ≥30 kg/m2; cystic fibrosis; or pulmonary malignancy.
Study treatments
Treatment consisted of aerosol therapy solely with BAY41-6551 400 mg b.i.d. in addition to standard i.v. antibiotic therapy for Gram-negative pneumonia and any other necessary interventions. Aerosol therapy was administered only after review of individual patients' serum creatinine levels obtained within 12 h before the start of the first dose of study medication on each day. In addition, trough serum amikacin concentrations (obtained daily approximately 30 min before the first scheduled dose) were reviewed before administration of the second daily dose. Patients with trough concentrations >5 mg/L did not receive the next scheduled dose; study drug administration was resumed only when the concentration was ≤5 mg/L.
Patients' i.v. antibiotic regimens were selected and monitored by the study site team, with the proviso that i.v. amikacin was prohibited during the aerosol treatment period, and that concomitant or sequential nephrotoxic products, particularly cisplatin, amphotericin B, cephaloridine, viomycin, polymyxin B, colistin, vancomycin, or other aminoglycoside antibiotics, and concomitant potent diuretics were to be avoided where possible.
Amikacin sulphate for inhalation was supplied as a specially formulated sulphite-free solution in vials containing 500 mg per vial in 4.0 mL of sterile water (concentration 125 mg/mL). Doses of 400 mg (3.2 mL) were administered every 12 h for 7–14 days. Aerosol treatments were administered using the proprietary PDDS Clinical on-vent device, in which the high-frequency OnQ vibrating plate provided breath-synchronized aerosol generation during the first 75% of each inspiratory cycle.(13) The duration of administration depends on the patient's minute ventilation; for a 3.2 mL (400 mg amikacin) nebulizer load, the delivery time is approximately 45–60 min.
For patients successfully extubated during the study treatment period, the PDDS Clinical hand-held device was substituted to provide continuously generated aerosol; administration takes approximately 15–20 min for a 3.2-mL dose.
Procedures and assessments
On day 3, the CVVHDF parameters extracorporeal blood flow, ultrafiltrate flow, and dialysate flow were recorded. Prisma® M100 Pre-Pump Infusion Sets (Gambro Lundia AB, Lakewood, CO, USA) were used in all patients. BUN was measured within 12 h of the first scheduled daily dose. At screening and follow-up, samples were obtained for clinical laboratory analysis (blood chemistry, hematology, and urinalysis).
Samples obtained on day 3 for PK analysis (timings relative to the first daily dose) included cumulative 24-hour urine (obtained in 12 hourly collections); blood obtained immediately before and at 0.5, 1, 3, 6, 9, 12, 13, and 24 h; and a BAL sample taken 15–30 min after completion of the morning dose with radiological guidance to obtain ELF from the infected area. Volume of ELF was estimated using urea as an endogenous marker of ELF dilution.(14) Blood samples for assessment of serum creatinine and trough amikacin concentrations were obtained approximately every 12 h during the study drug treatment period.
PK samples obtained on day 3 (serum, urine, and BAL fluid) were analyzed by the central laboratory (MEDTOX Laboratories, Saint Paul, MN, USA). Amikacin in serum was assayed using a high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS)-based method using electrospray spectrometry conditions on an ABI-Sciex 5000 triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA, USA). Amikacin in urine and BAL samples was assayed using an enzyme-multiplied immunoassay Syva® EMIT® kit (Siemens Healthcare Diagnostics, Deerfield, IL, USA). These methods, which have been reported previously,(14) were prevalidated according to current U.S. Food and Drug Administration guidelines. The lower limits of quantification of the assays used to determine amikacin concentrations were 200 μg/L in serum and 2.5 mg/L in both urine and ELF. Assay accuracy ranges validated from standard curves were 98–110% for serum, 94–98% for urine, and 91–101% for ELF.
Daily serum creatinine and trough amikacin concentrations were determined by the local laboratory. Creatinine serum levels were measured by a colorimetric method (Jaffe method) on an automatic analyzer (Cobas®, Roche Diagnostics Ltd, Basel, Switzerland). The lower limit of quantification of the assay was 18 μmol/L. Intra- and interindividual variations were 0.7 and 2.3%, respectively. Daily trough amikacin concentrations were measured using the fluorescence polarization immunoassay method (Abbott Laboratories, Abbott Park, IL, USA). The lowest measurable concentration that can be distinguished from zero with 95% confidence was determined to be 0.8 mg/L. Intra- and interindividual variations were below 5%. The accuracy of the method, assessed by correlation with reference assays (radioimmunoassay and substance-labeled fluorescence immunoassay), was 96.5–97.7%.
Serum amikacin concentration values obtained on day 3 were used to calculate maximum serum drug concentration (Cmax), time to Cmax (tmax) and area under the serum concentration–time curve (AUC) from zero to 12 h (AUC12) using noncompartmental analysis. Urine obtained on day 3 was used to calculate the cumulative amount of amikacin excreted (Ae) at 0–12, 12–24, and 0–24 h after administration of the first dose of study drug of the day. ELF amikacin concentrations were determined from BAL samples obtained on day 3.
Study objectives
The primary objective was determination of the PK profile of aerosolized amikacin following administration of multiple doses of BAY41-6551. Characterizing the safety and tolerability profile of BAY41-6551 was a secondary objective. Medical Dictionary of Regulatory Activities (MedDRA) version 9.1 was used to assign system organ classes and preferred terms to AE verbatim terms.
Analysis populations
Two populations were defined for analysis purposes: the safety population, comprising all enrolled patients who received study treatment; and the PK population, comprising all treated patients who had evaluable PK data for any of the specimen types obtained on day 3 and who received at least 3 full days of study medication.
Statistical methods
A target sample size of six patients was planned to enable adequate qualitative clinical assessment of safety, tolerability, and PK. Results are presented as descriptive summary statistics. All analyses were conducted using SAS® (Cary, NC, USA) version 8.2 or higher on a PC platform.
Results
Study sample and extent of BAY41-6551 exposure
Seven patients were enrolled: four completed the study and three terminated before completing the follow-up visit. The safety population comprised all seven patients. The PK population (n = 6) excluded one patient who died on day 2. For all patients, quantitative bacteriological cultures confirmed the diagnosis of pneumonia. Demographic and baseline clinical characteristics of the enrolled patients are summarized in Table 1. The mean [standard deviation (SD)] total duration of BAY41-6551 treatment in the safety population was 6.29 (1.89) days. Six patients were treated for 5–8 days and one patient was treated for 2 days. The mean (SD) numbers of BAY41-6551 doses per patient administered via the on-vent and hand-held PDDS Clinical devices were 11.00 (4.28) and 4.00 (4.24), respectively. Two patients in the safety population were extubated during the study period: one received a single BAY41-6551 administration via the PDDS Clinical hand-held device and the other received seven administrations via the PDDS Clinical hand-held device.
ARDS, acute respiratory distress syndrome; BMI, body mass index; FIO2, fraction of inspired oxygen; MV, mechanical ventilation; PaO2, alveolar partial pressure of oxygen; SD, standard deviation; WBC, white blood cell.
Systemic amikacin PK
Individual patient and geometric mean 24-h serum concentration–time profiles on day 3 are shown in Figure 1 (central laboratory data). Mean [standard error (SE)] and median (range) values on day 3 for serum amikacin Cmax were 2.20 (0.57) mg/L and 1.93 (0.63–3.99) mg/L, respectively; for AUC12 were 20.49 (5.04) mg · h/L and 19.32 (6.32–36.87) mg · h/L, respectively; and for tmax were 2.42 (0.38) h and 3.00 (1.00–3.00) h, respectively. Only one patient provided urine for the assessment of Ae on day 3: total 24-h Ae was 52.7 mg (18.7 mg for the 0- to 12-h collection and 34.0 mg for the 12- to 24-h collection). Daily serum trough amikacin concentrations are summarized in Figure 2 (local laboratory data).
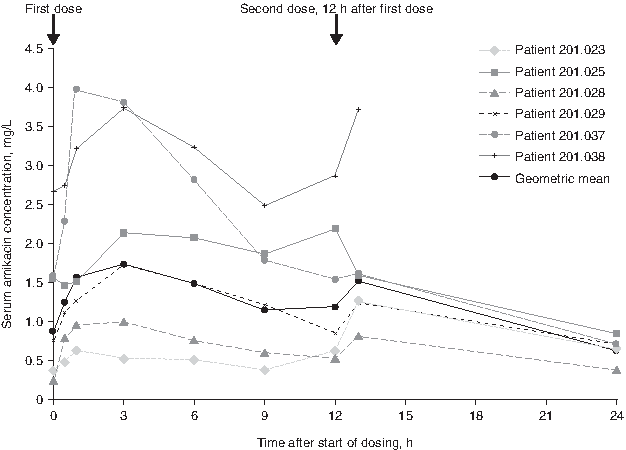
Individual patient and geometric mean amikacin serum concentration–time profiles on day 3 as assayed by HPLC-MS/MS (pharmacokinetic population; N = 6). The first sample taken after the second dose was at 1-h postsecond dose completion, with the exception of patients 201.025 and 201.038, who were sampled at the beginning of the administration of the second dose and patient 201.023, who was sampled 30 min after.
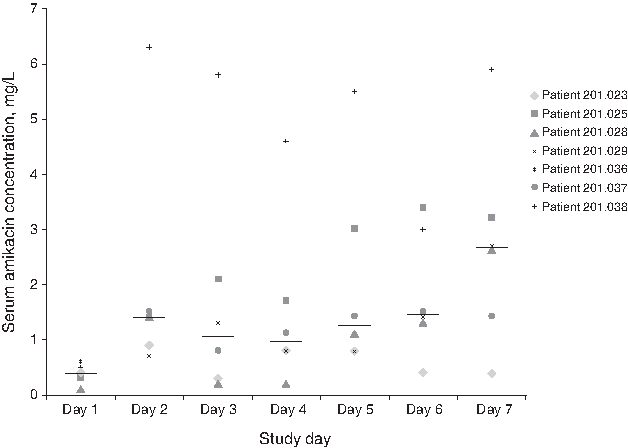
Dot-plot of trough serum amikacin concentrations assayed by fluorescence polarization immunoassay (safety population; N = 7). Daily serum amikacin trough concentrations were measured 30 minutes prior to first dosing. The horizontal lines represent median values.
ELF amikacin concentrations
The mean (SE) and median (range) amikacin concentration in ELF (PK population) were 2867 (1996) mg/L and 887 (406–12,819) mg/L, respectively. The range of peak inspiratory flow rates was relatively narrow; however, there did not appear to be a relationship between peak inspiratory flow rate and amikacin ELF concentration.
Ventilator parameters
No patients receiving mechanical ventilation required changes in their ventilator settings during the study treatment period. All patients used the ventilator in assist control mode; five patients also used other ventilator modes.
CVVHDF parameters
Mean (SD) CVVHDF parameter values on day 3 (PK population) were 2000 (0) mL/h for ultrafiltrate flow and 2083 (204) mL/h for dialysate flow. Extracorporeal blood flow was 10,700 (0.96) mL/h.
Clinical laboratory parameters
Six of the seven patients in the safety population had abnormal elevations in their white blood cell counts at screening (Table 1), which had decreased by early withdrawal or follow-up in three patients. Five patients had clinically significant abnormal BUN elevations during the study.
Safety and tolerability
Mean serum creatinine levels (safety population) fluctuated between approximately 0.9 mg/dL and 1.5 mg/dL over the course of the study with no apparent trend; serum creatinine was reported as abnormal in six patients, with clinically significant elevations reported in five patients. A total of 119 adverse events (AEs) were reported; most (89.9%) were as expected for the patient population. The most frequently reported expected AEs were anemia, hyperglycemia, leukocytosis, and purulent respiratory secretions (7/7 patients).
A total of 12 unexpected AEs were reported in six patients (Table 2). Of these, three events in three patients were classified as serious AEs (SAEs): one episode of multiorgan failure (MOF), one of worsening MOF, and one of bronchospasm. The patient with worsening MOF died on day 2 after initial intensive care unit admission for cardiac arrest secondary to ventilator-associated pneumonia. A second patient with MOF secondary to pneumonia and cardiogenic shock died on day 30 (23 days after completing 7 full days of study medication). The MOF events were considered unrelated to the study medication. The third SAE, bronchospasm, was initially reported on day 6. BAY41-6551 was discontinued, the patient was treated with a nebulized β-2 agonist and the event resolved. Before resuming study medication, the patient was pretreated with a β-2 agonist, but experienced a second episode of severe bronchospasm within 15 min of administration of BAY41-6551. Manual ventilation and remedial drug therapy for a profound decrease in oxygen saturation with bradycardia were instituted. BAY41-6551 was discontinued permanently; the event resolved and was considered probably related to the study medication. No reported AEs were considered to be related to the PDDS Clinical device.
The total number of AEs counts all reported events for all patients. At each level of summarization, a patient is counted only once if the patient reported one or more events.
Discussion
This study of adjunctive BAY41-6551 treatment in intubated and mechanically ventilated patients with Gram-negative pneumonia and ARF supports the expected relationship between renal function and systemic amikacin exposure following BAY41-6551 aerosol treatment. It is the first prospective evaluation to our knowledge of systemic PK following inhaled aminoglycoside administration in patients receiving renal replacement therapy for ARF. Median serum amikacin AUC12 on day 3 was three to four times greater compared with mechanically ventilated patients with normal renal function in a study of identical design except for the inclusion of patients with ARF,(14) and the serum amikacin concentration–time profile was characterized by a prolonged mean/median tmax and elevated mean/median Cmax. The observation of increased systemic exposure in patients with ARF is consistent with findings from a single-dose PK study in subjects with various degrees of renal insufficiency.(15)
In the study of patients with normal renal function, the median (interquartile range) serum Cmax and tmax were 0.85 (0.67–1.01) mg/L and 1.00 (1.00–3.00) h, respectively; AUC12 was 6.15 (4.73–9.57) mg.h/L.(14) In patients with ARF, although the respective parameters were elevated, serum amikacin concentrations measured by the central laboratory were <4 mg/L at all times, well below the maximum recommended trough concentration (i.e., <10 mg/L) for i.v. amikacin.(16) Furthermore, the 24-h Ae value of 52.7 mg from the one patient who produced urine on day 3 is consistent with the relatively low overall systemic amikacin exposure indicated by the serum results. CVVHDF thus appeared to provide adequate clearance of systemically absorbed amikacin, suggesting that BAY41-6551 dose adjustments are probably not necessary for this patient population.
Direct comparisons cannot be made between serum concentrations measured by the local laboratory (using a fluorescence polarization immunoassay) and those measured by the central laboratory (using a HPLC-MS/MS-based method): measures of trough serum amikacin measured by the former were in one case higher than the Cmax amikacin values measured by the latter, a discrepancy possibly due to differences in the methodology. It is believed that immunoassays may be associated with reduced accuracy;(17,18) a previous study of BAY41-6551 adopted a HPLC-MS/MS-based method for the evaluation of serum amikacin concentration because of the assay's sensitivity.(14) Nevertheless, serum amikacin concentrations in all patients at any time points and by either method were always below the maximum recommended trough concentration (10 mg/L) for i.v. amikacin.(11,16)
In this study, the median day 7 serum amikacin trough concentration was greater than that at day 4. Given the low patient numbers, it is difficult to comment on any clinical significance of this, but it does suggest that there may be a potential for systemic accumulation of amikacin in individual patients with ARF over time, and underlines the need for continued monitoring of serum amikacin concentrations in such patients receiving BAY41-6551.
Both in patients with ARF and in those with normal renal function, BAY41-6551 achieved very high amikacin concentrations in ELF samples from areas of pneumonic lung tissue; values obtained from patients with ARF were broadly similar to the median (range) of 976.07 (135.67–16,127.56) mg/L reported for patients with normal renal function.(14) These values are one to three orders of magnitude higher than mean amikacin concentrations in the bronchial secretions of patients with pneumonia who received i.v. amikacin in a historical study.(11) Although the observed ELF concentrations demonstrated substantial interpatient variability, the lowest recorded concentrations were >15–20 times the European Committee on Antimicrobial Susceptibility Testing susceptible breakpoint of 8 mg/L for amikacin against Pseudomonas aeruginosa,(19) one of the most problematic Gram-negative pathogens, and the median value was >100 times the 8 mg/L susceptible breakpoint.
BAY41-6551 demonstrated adequate safety and tolerability in this study. The majority of reported AEs were expected, given the study population of critically ill patients; of the 12 unexpected AEs, one (an SAE of bronchospasm) was considered related to the study drug and none was related to the PDDS Clinical device. Because investigators were instructed to avoid concurrent nephrotoxic treatments and potent diuretics, the study provides limited data on the concurrent use of BAY41-6551 with many interventions frequently used in the critical care setting. Furthermore, with this small study population, the safety of adjunctive BAY41-6551 alongside the multiple combinations of i.v. antibiotic regimens available for treating pneumonia could not be comprehensively assessed. Consequently, the standard precautionary measures for critically ill patients receiving i.v. amikacin (including assessments of renal function and monitoring of therapeutic drug levels and AEs) should be followed for patients with ARF treated with BAY41-6551. Physicians should consider withholding repeat BAY41-6551 doses if trough amikacin levels are above a defined threshold (usually 10 mg/L for systemic administration), and caution should be exercised when concomitant administration of other potentially nephrotoxic treatments is deemed necessary.
A further limitation of this study is the determination of amikacin ELF concentrations from single BAL samples. In this respect, the study design is constrained by ethical considerations for serial invasive procedures.(20) It is likely that the wide interpatient variation in amikacin ELF concentrations observed in both this and the identical study of patients with normal renal function was partly due to the inherent limitations of bronchoscopic sampling techniques, as well as intraindividual variability in pulmonary deposition; variability in airway deposition might also account for the intraindividual serum amikacin variability seen in both studies. Despite these difficulties in interpreting pulmonary drug deposition data, the study achieved its aim of characterizing systemic amikacin PK in patients with ARF following administration of multiple doses of BAY41-6551. Phase III, superiority-design, randomized prospective studies will evaluate the clinical efficacy of adjunctive BAY41-6551 400 mg b.i.d. in combination with standard-of-care i.v. antibiotics in mechanically ventilated patients with Gram-negative pneumonia.
Footnotes
Acknowledgments
Medical monitoring of this study was conducted by David Sahner, M.D., and the study safety medical officer, Sunita Dhar, M.D., of Nektar Therapeutics. Overall study management, data management, and statistical analyses were performed by PPD Inc (Wilmington, NC, USA). The authors acknowledge Magdalene Chu of Chameleon Communications International Ltd., who provided medical writing assistance with funding from Bayer Schering Pharma.
Author Disclosure Statement
Kevin Corkery and Dennis Gribben were employed by Nektar Therapeutics during the conduct of the clinical trial. As part of compensation of employment, Kevin Corkery received stock. Heino Stass is an employee of Bayer Schering Pharma AG. Jean Chastre has provided consultancies and received honoraria from Bayer Schering Pharma AG and Nektar Therapeutics.