
Abstract
Abstract
Background:
This report presents results of the first human study of a new dry powder inhaler (DPI-C). DPI-C uses reverse flow cyclone technology to retain larger particles in the device and to increase efficiency of respirable drug release. The study was conducted to determine comparative pharmacokinetics (not bioequivalence) of DPI-C and DPI-A (Advair Diskus®, GlaxoSmithKline) and to establish preliminary efficacy and safety of DPI-C.
Methods:
Nineteen patients with mild–moderate asthma received two treatments (randomized crossover design). Treatments were one inhalation from DPI-A labeled to deliver 100 μg fluticasone propionate and 50 μg salmeterol, or one inhalation from DPI-C which contained ∼10% less of each drug per metered dose. Prior to dosing, 10 g of charcoal was administered. FEV1 increase over baseline (measured over 12 h), plasma concentrations of fluticasone and salmeterol (measured over 12.5 h), and occurrence of adverse events were the primary measures of device performance and safety.
Results:
Seventeen patients were evaluable. Response profiles of percent increase in FEV1 over baseline showed no statistically significant differences between devices. Peak plasma concentrations of both fluticasone (p = 0.003) and salmeterol (p = 0.084) were higher from DPI-C. Mean extent of absorption [area under the curve (AUC)] of fluticasone was approximately 30% greater with DPI-C, whereas AUC of salmeterol was approximately 40% greater with DPI-A.
Conclusions:
DPI-C provided similar improvement in pulmonary function compared with DPI-A. Pharmacokinetic results showed a greater initial absorption of salmeterol with DPI-C but greater continued absorption and a 40% greater AUC with DPI-A, which we attribute to slower but more extensive oral absorption because of the greater mass of swallowed large particles of salmeterol generated by DPI-A. No patient reported any treatment-related adverse event or use of rescue medication during this study. Determination of the significance of the observed differences in pharmacokinetics from this single-dose study requires further exploration in studies using clinically relevant dosing regimens.
Introduction
DPI devices can be categorized into four distinct types. Devices are either active or passive, and utilize either a reservoir of formulation that is sampled by the device to meter a dose, or premetered doses.(3) All combinations of these two categories are potential options to the product developer, each having its own strengths and challenges. Historically, devices reaching the market have typically been passive systems due to the unit cost and regulatory challenges associated with active devices; however this is now changing and active devices are starting to reach the market. Both reservoir (e.g., Turbuhaler™) and premetered (e.g., Handihaler™) devices are well represented in the marketplace.
DPI technology is still relatively new and is undergoing improvements. 3M has focused on perceived needs of new DPI devices to offer improved robustness, higher efficiency, increased respirable fraction, and decreased swallowed drug. To meet these needs, the Conix™ Dry Powder Inhaler (DPI-C) was developed. This device uses an innovative reverse-flow cyclone design to offer effective drug delivery and simple operation.
This report presents the results of the first study of DPI-C in humans. The purpose of this study was to determine the capability of this device to effectively and safely deliver a combination therapy (fluticasone propionate and salmeterol xinafoate) to asthmatic patients. The reference chosen was the marketed product of the same combination, Advair Diskus® (DPI-A).
Reverse flow cyclone technology
Most dry powder inhaler devices utilize a blend of micronized drug and coarse carrier particles, typically lactose, to add bulk to the formulation. This approach is intended to make the metering and delivery of the drug more reproducible. DPI devices are designed to deagglomerate the lactose/drug particles through application of shear forces or induction of particle/particle and particle/surface collisions.(4) The design intent of the Conix technology is to retain larger particles in the device so as to continue to expose them to these deagglomeration processes to increase the efficiency of the respirable drug release. Any drug leaving the device while still agglomerated with lactose particles will subsequently impact on the patient's throat and only the smaller drug particles are delivered to the lung.
In the DPI-C device, deagglomeration and aerosolization of the drug occur through reverse-flow cyclone technology (Fig. 1). As the patient inhales, air is drawn into the cyclone chamber, where a vortex is established. The base of the vortex cone is blocked such that when the vortex hits the bottom, the flow reverses and is forced through the center of the incoming air towards the exit orifice. This is known as a forced vortex.
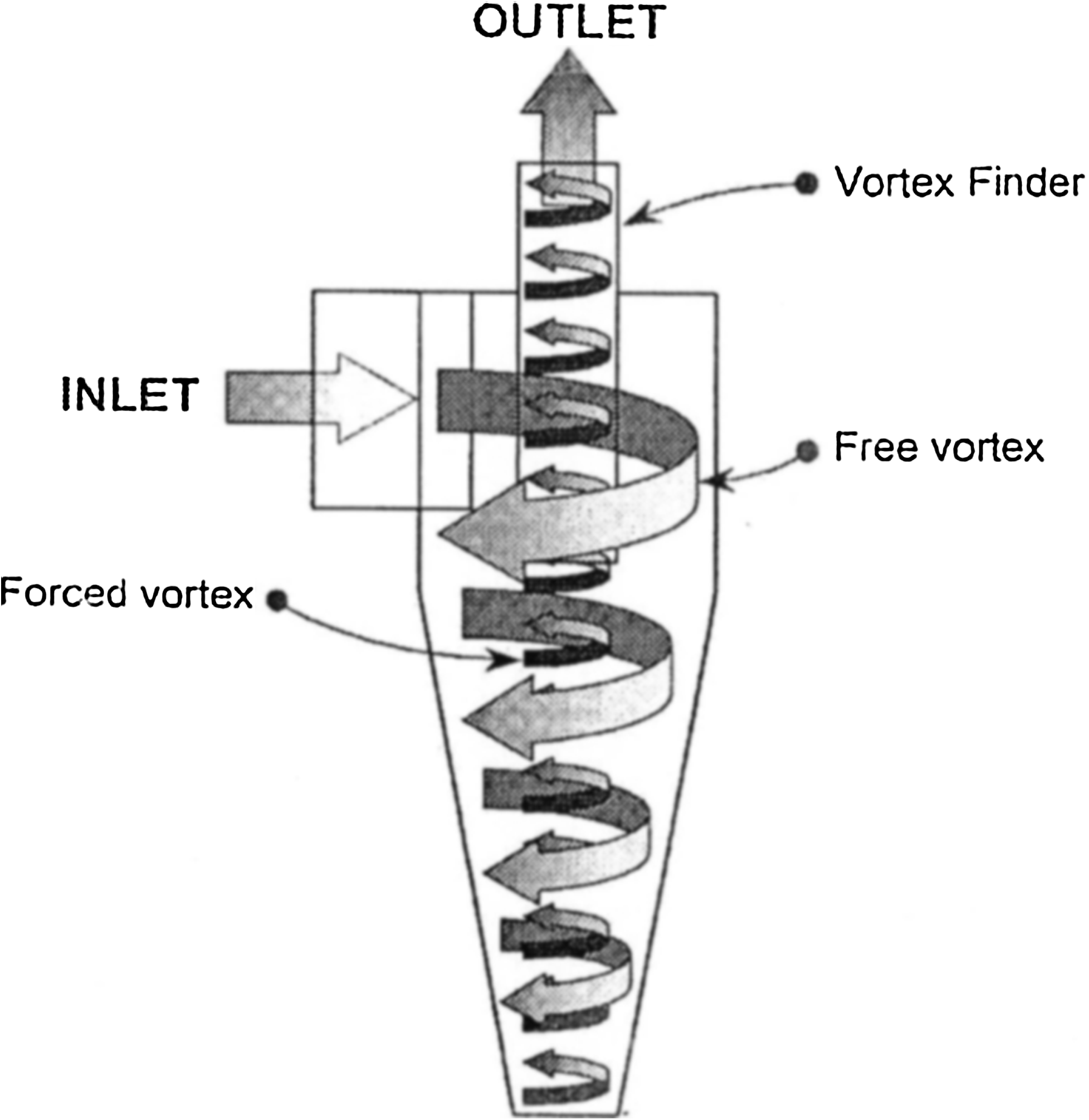
DPI-C reverse flow cyclone technology. Incoming air enters the cyclone chamber in a downward spiral (free vortex), is forced to reverse direction at the sealed bottom while maintaining rotation (forced vortex), and exits at the top through the vortex finder.
The vortex produced by the reverse flow cyclone creates relatively high velocities and therefore energy in the air flow, which is required for deagglomeration through collisions with the cone wall, collisions with other particles, and through particle shear. As deagglomeration occurs, the large lactose particles are flung to the sides of the cyclone and the lighter drug particles are carried along by the air flow and exit the cyclone. Therefore, any drug still adhered to lactose particles is retained in the deagglomeration process rather than being emitted, typically to impact in the throat, thus maximizing the likelihood of drug becoming respirable to the user.
Materials and Methods
Materials
DPI-A (Advair Diskus®, GlaxoSmithKline, Research Triangle Park, NC) and activated charcoal (Actidose®, Paddock Laboratories, Minneapolis, MN) were purchased from commercial sources. DPI-C was manufactured at 3M Drug Delivery Systems (Loughborough, UK). The amounts of drugs in each inhalation dose from DPI-C were similar but slightly lower than those of each inhalation dose from DPI-A (Table 1). The same DPI lots were used for the in vitro and in vivo studies.
The dose in the DPI-C is contained within a foil-covered blister. Upon activation, the foil is pierced to expose the drug. Due to development delays, the final step of piercing the foil could not be validated prior to the scheduled start of the clinical study. Rather than postpone the study, the foil was manually removed by the clinician since this action had no effect on the cyclone mechanism that differentiated the device.
DPI-C Inhalation formulation
As much of the information needed to fully characterize the starting materials of the innovator was commercially sensitive and not freely available, no attempt was made to match the primary characteristics of the drugs and excipient of the innovator. Our approach to match the innovator formulation had to be pragmatic. The basic formulation type was mirrored, that is, the same strengths of the two drugs in a lactose carrier were utilized. The DPI-C inhalation formulation of fluticasone propionate and salmeterol xinafoate was developed to be comparable to the in vitro respirable particle dose of each drug from the innovator product DPI-A.
Although particles of 5 μm are likely to be respirable, there may not be a good correlation with lung deposition of the drug.(5,6) The optimum size for deposition into the smaller lung spacers and clinical efficacy appears to be in the region of ≤3 μm; hence, this size range was selected for in vitro matching of the devices.(6) The total masses of particles ≤3 μm of each drug delivered from DPI-C were successfully matched (within ± 15%) to those from DPI-A using Next Generation Impactor (NGI) testing and high-performance liquid chromatography (HPLC) analysis (Table 2). DPI-C was able to achieve a comparable particle mass ≤3 μm with only about one-third of the delivered dose of DPI-A, a major benefit of reverse flow cyclone technology.
Delivered dose derived from total mass of drug on impactor (amount delivered exmouthpiece).
Although the in vitro particle sizing conditions used to determine the fine particle dose may be arbitrary and clinically irrelevant, they conform to the compendial (USP) testing method for Apparatus 5 (NGI) for a DPI, which specifies a total volume of 4 L and a constant pressure drop of 4 kPa.(7) To maintain 4 kPa, the, flow rates of the NGI tests were approximately 63 L/min for DPI-C and 79 L/min for DPI-A.
The DPI-C device released far fewer large particles per actuation than did DPI-A, which was seen in the NGI profiles as less preseparator and throat deposition for each delivered drug (Fig. 2). Because the significant differences in the generation of large particles between the products suggests possible differences in the amount of drugs swallowed in vivo, an oral charcoal pretreatment was included in the study.
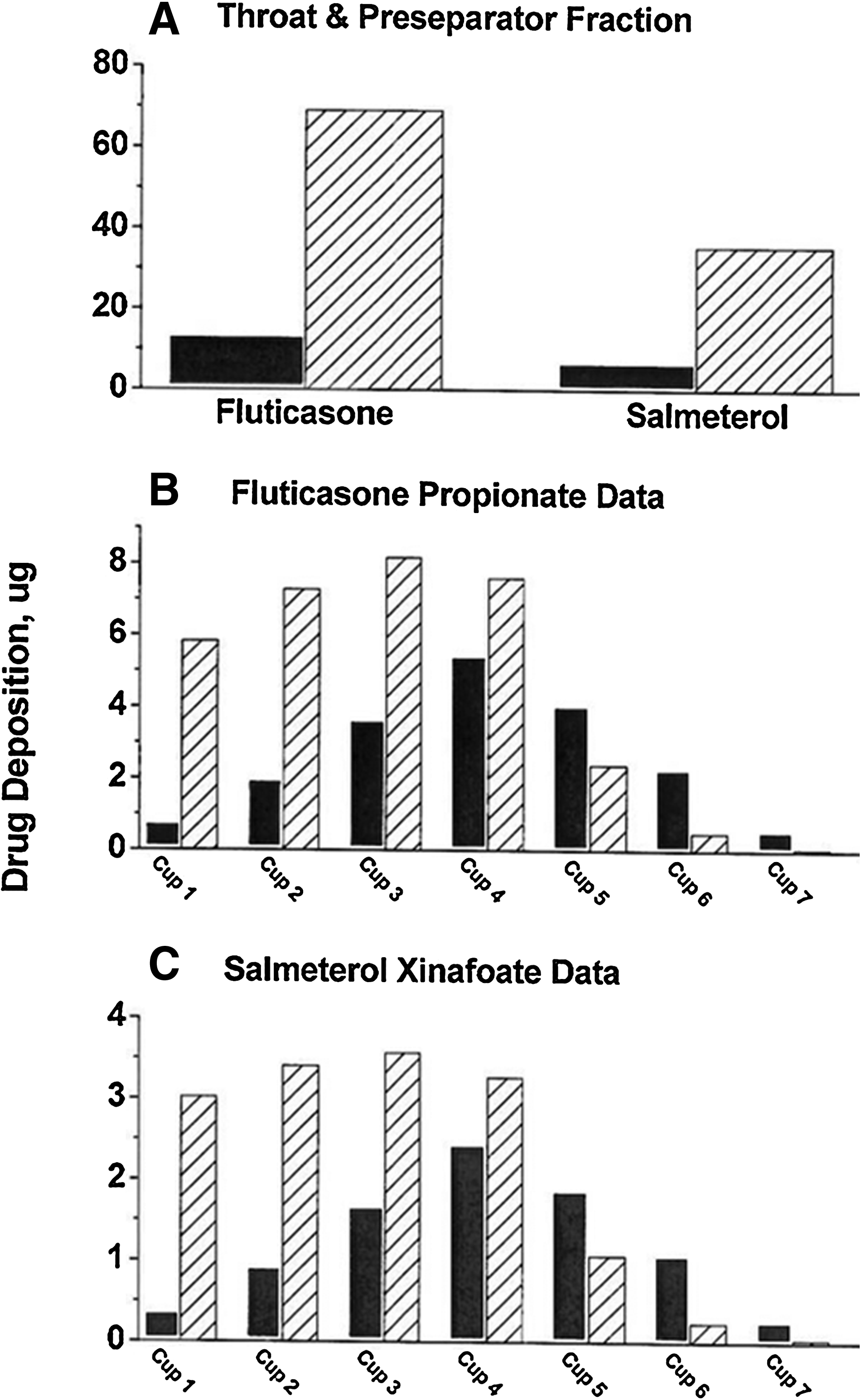
Comparison of NGI profiles for fluticasone propionate and salmeterol xinafoate from DPI-C (solid bar) and DPI-A (hatched bar). Particle collection on the last stage (micro orifice collector) was 0.3 μg for all analyses; data not shown.
Clinical study
Patients
Nineteen adult patients with mild-to-moderate asthma (FEV1 of 50–80% of predicted), who had been stable for at least 6 months and who were otherwise healthy, were enrolled. Patients had to demonstrate at least 12% reversibility of bronchoconstriction with two inhalations of albuterol sulfate. Female patients had to be using an accepted method of birth control and could not be pregnant or nursing. Patients were excluded if they had a history of drug or alcohol abuse within the past 2 years, or smoked tobacco within the past 12 months, or had a history of more than 10-pack years.
Ethical review
All patients gave written informed consent before they were included in the study and the protocol was approved by an institutional review board.
Supplies
DPI-C was manufactured by 3M Drug Delivery Systems (Loughborough, UK). DPI-A (GlaxoSmithKline, Research Triangle Park NC) and activated charcoal (Actidose®, Paddock Laboratories, Minneapolis, MN) were purchased from commercial sources.
Design
The study was a single-center, open-label, trial conducted by Sylvana Research (San Antonio, TX). All patients had to demonstrate correct inhalation technique with placebo inhalers of both products. Immediately prior to the study treatment days, patients abstained from their current maintenance asthma medication for at least 24 h, food and fluids other than water for 12 h, and the use of any rescue asthma medication for at least 6 h. On the treatment days, patients' FEV1 values had to be ≤80% of predicted to continue. Two minutes prior to each treatment, 10 g of activated charcoal was administered in 100 mL of water. The patient swirled the charcoal in his/her mouth before swallowing. No additional water or food was allowed until 1 or 2 h postdosing, respectively.
The reference treatment was one inhalation from DPI-A labeled to deliver 100 μg fluticasone propionate and 50 μg salmeterol base equivalent. The test treatment was one inhalation from DPI-C which contained approximately 10% less of each drug per premetered dose in a lactose blend. Patients received the two treatments separated by 7–16 days, according to a randomized, two-period crossover design. Between treatment days, patients resumed their prescribed asthma medication.
Spirometry
On treatment days, pulmonary function was measured predose, at 30 min, and hourly for 12 h postdosing using a calibrated spirometer. All spirometry was performed with the patient in a sitting position. Testing was performed in compliance with the ATS standards.(8) The patient was instructed to give a maximal forced expiration over at least 6 sec, or for 3 sec with at least a 1 sec plateau. Three or more measurements were made. FEV1 was the primary parameter. The value recorded for FEV1 was the highest value obtained from three acceptable curves.
Pharmacokinetics
On treatment days, blood samples were collected at 0, 5, 10, 15, 20, 30, and 45 min, and at 1, 1.5, 2.5, 4.5, 6.5, 8.5, 10.5, and 12.5 h for pharmacokinetic analyses. Plasma was collected using potassium oxalate/sodium fluoride anticoagulant. All drug plasma analyses were done with proprietary validated LC/MS/MS methods by PPD (Middleton, WI). The lower limit of sensitivity for each drug was 1 pg/mL.
Safety
Safety was assessed for serious and nonserious adverse events, from the time of the signing of the informed consent through a follow-up phone call at 30–36 days posttreatment. The use of concomitant medications was also recorded.
Statistical analyses
The dataset analyzed consisted of all randomized patients who completed the two treatment periods. The primary efficacy response was the percent change from predose FEV1 values. The FEV1 response values were analyzed using a linear mixed effects model with restricted maximum likelihood estimation. Dosing sequence, treatment (DPI-A or DPI-C), and period were fixed effects, while patient nested within sequence was a random effect. Baseline FEV1 values for each patient were added to the model as a covariate. Peak response (Rmax) and area-under-the-response versus time curve (AUCR) were the primary FEV1 responses. The pharmacokinetic responses were analyzed with the same linear mixed effects model, but did not include the covariate. Peak concentration (Cmax), and area-under-the-plasma concentration versus time curve (AUC) were the primary pharmacokinetic responses. All responses were transformed prior to statistical analysis by computing natural logs. The effect of treatment on all responses was evaluated for statistical significance (α = 0.05).
Results and Discussion
Demographics
Eleven males and eight females were enrolled, with an average age of 33 years (range: 18–54 years). Their FEV1 averaged 72% of predicted (range: 59–79%). Eighteen patients completed the study and one patient withdrew after one treatment period for personal reasons. One patient was not included in the pulmonary function analyses because FEV1 varied by more than 20% on the two study days; however, because this was an exploratory study, the protocol allowed patients who were noncompleters for pulmonary function analyses to be included in the pharmacokinetic analyses. This decision was supported by the finding that fluticasone pharmacokinetics did not correlate with FEV1.(9) Similarly, an initial patient was not included in the pharmacokinetic analyses because of predose sample contamination of both analytes, but was included in the pulmonary function analyses; this contamination appeared to be a sample handling issue that was discovered following the analysis of the first four patients' samples. It was corrected and did not occur again. Thus, the FEV1 responses for 17 patients were analyzed, and the pharmacokinetics responses for 17 patients were analyzed.
FEV1
The response profiles of the percent increase in FEV1 over baseline were essentially the same for the two treatments (Fig. 3). The derived FEV1 pharmacokinetic parameters are presented in Table 3. No statistically significant differences (α = 0.05) between the treatments were seen in any of the parameters in Table 3.

Percent increase in FEV1 over baseline, mean ± SEM (N = 17), following administration of the DPI-C inhaler (solid squares) and the DPI-A (open circles).
Units: TRma
Data presented are mean ± SD (N = 17).
p-Values generated from Proc Mixed in SAS® 9.2 on data after natural log transformation.
The time of Rmax (TRmax) varied widely and unpredictably over the observation interval between treatment days among the patients and did not appear to be a useful parameter. Less variability was seen among the patients for Rmax and AUCR. Still, there were three patients given DPI-A that had an AUCR that was less than 20% of the mean, including one patient that had a net value of zero due to FEV1 values that fell below baseline during treatment; there were two patients given DPI-C in this range. Three patients given each treatment had an AUCR that was greater than 175% of the mean.
Pharmacokinetics of fluticasone
Fluticasone was absorbed faster and more extensively from DPI-C than from DPI-A. The differences were large over the first 2.5 h (Fig. 4). Cmax was more than double and AUC approximately 30% greater, on average, following DPI-C. Statistically significant differences between the treatments were seen for Cmax and Tmax (Table 4).
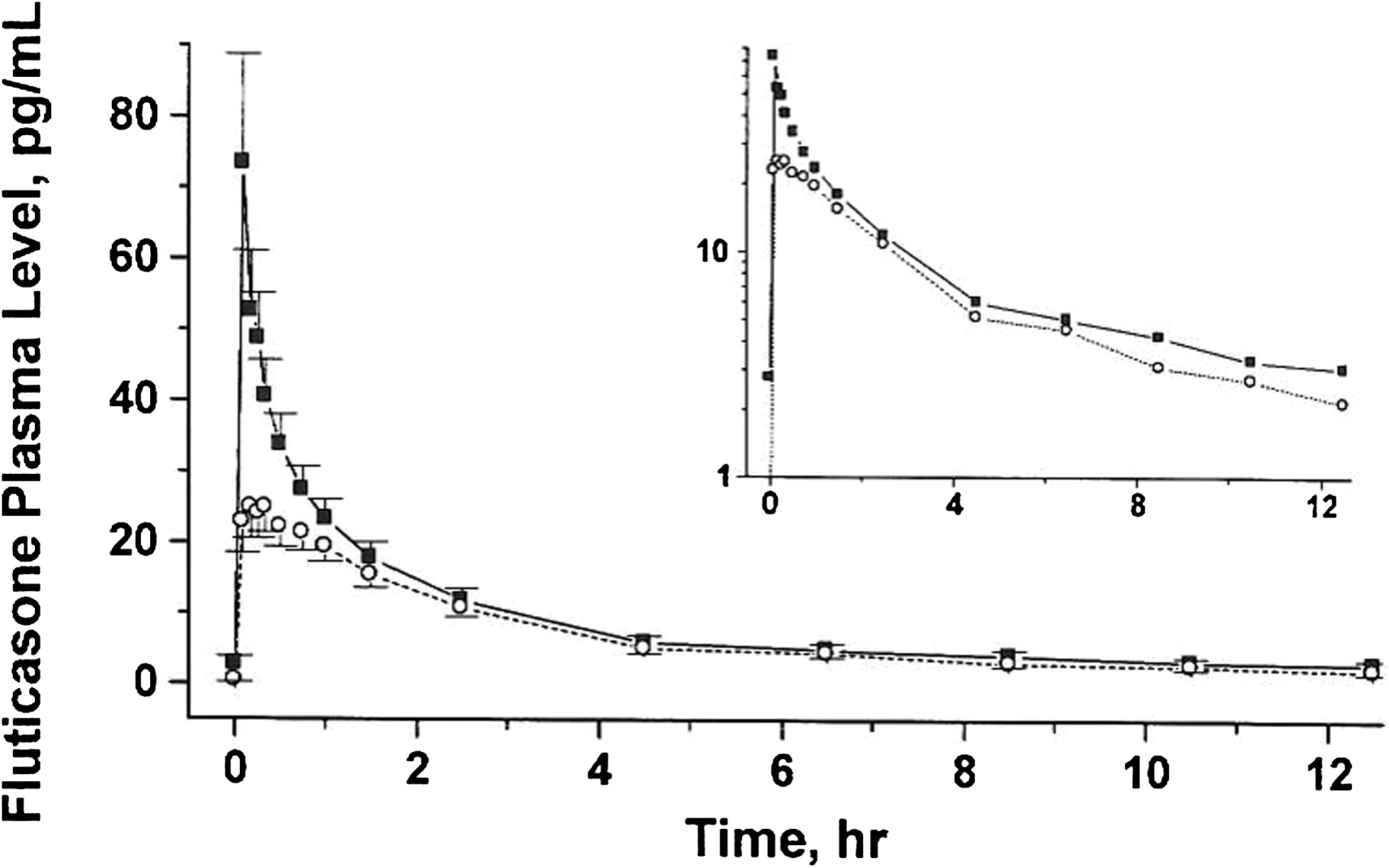
Fluticasone plasma levels, mean ± SEM (N = 17), following administration of the DPI-C inhaler (solid squares) and the DPI-A (open circles). The insert is a semilog plot.
Units: Tma
Data presented are mean ± SD (N = 17).
p-Values generated from Proc Mixed in SAS® 9.2 on data after natural log transformation.
It has been reported that the oral and buccal absorption of fluticasone is minimal, estimated to be less than 1%.(10) The plasma profiles in Figure 4 therefore represent the systemic absorption of the fraction of fluticasone deposited in the lungs. Previous studies over a large age range of asthmatic patients have established that the pulmonary absorption of small particles is faster and more extensive than that of large particles.(11,12) The current results are in agreement with this observation. The in vitro experiments showed that the DPI-C inhaler generated a larger fraction of small particles of fluticasone than did DPI-A, as evidenced by the higher amounts in cups 5–7 of the NGI (Fig. 2) and a lower MMAD. In vivo, this difference in the amount of small particles appeared to result in an initial faster absorption rate of fluticasone that produced a higher Cmax and a 30% greater AUC for DPI-C (Fig. 4, insert).
Pharmacokinetics of salmeterol
Salmeterol was initially absorbed faster from DPI-C but absorption continued longer and more extensively with DPI-A (Fig. 5). It was apparent that DPI-A maintained higher plasma levels of salmeterol over most of the observation interval, which resulted in a 40% higher AUC, on average, than did DPI-C. The large intrapatient variability in plasma levels of this analyte prevented any pharmacokinetic parameter from showing statistical significance (Table 5).

Salmeterol plasma levels, mean ± SEM (N = 17), following administration of the DPI-C inhaler (solid squares) and the DPI-A (open circles). The insert is a semilog plot.
Units: Tma
Data presented are mean ± SD (N = 17).
p-Values generated from Proc Mixed in SAS® 9.2 on data after natural log transformation.
For all of the subjects given DPI-C and 14 of the 17 subjects given DPI-A, salmeterol Tmax occurred at 5 min, the first sampling time. Perhaps more intersubject variability might have been observed if blood sampling initiated sooner. However, the literature includes a report of another pharmacokinetic study of DPI-A and an experimental DPI that measured the plasma levels of salmeterol following 14 days of dosing at 2 min as well as 5 min.(13) That study reported a median Tmax of 5 min for both DPI-A and the experimental DPI. A salmeterol median Tmax of 5 min (range: 2–6 min) has been reported following four inhalations from of a commercial pressurized metered-dose inhaler (Seretide™) delivering fluticasone/salmeterol (250/25).(14) Thus, our finding of a salmeterol mean Tmax of 5 min is in agreement with that reported in the literature and is probably an accurate assessment.
Unlike fluticasone, it has been estimated that approximately one-third of the pharmacological activity following inhaled salmeterol comes from swallowed drug.(15,16) The interpretation of the salmeterol profiles in Figure 5 are therefore complex, reflecting both pulmonary and oral absorption. Based on the fluticasone data, it was assumed that the plasma levels for the first 30 min reflected primarily pulmonary absorption. The more rapid and higher plasma levels observed following DPI-C are consistent with the interpretation that this was due to the larger amount of smaller particles within the ≤3-μm fraction generated with this inhaler. Around 30 min, a major change was seen in the salmeterol pharmacokinetics. Whereas the plasma levels from the DPI-C treatments continue to fall as expected with pulmonary deposition, the salmeterol plasma levels from DPI-A showed a profile that was consistent with continued slow absorption that persisted for 4 h (Fig. 5, insert). Our supposition is that by 30 min, some swallowed salmeterol would have passed through the stomach and entered the small intestines and begun to be absorbed. Considering the transit time through the small intestines, it is conceivable that oral absorption of swallowed salmeterol could have persisted for 4 h.(17) The finding that this continued absorption was only observed following DPI-A was not surprising considering that the DPI-C inhaler was designed to generate fewer large particles and actually emitted a much smaller freaction of large particles (Fig. 2).
Because the focus of this study was to collect preliminary evidence for efficacy and safety (adverse event profile) of DPI-C, the simplest charcoal procedure was selected. The literature contains reports of the effectiveness of a single charcoal treatment in blocking oral absorption following inhalation of another beta-agonist (albuterol) and also of cromoglycate.(18,19) It appears that the single charcoal pretreatment employed in the present study was not sufficient to block the amount of swallowed salmeterol that occurred, and that the published methods requiring multiple charcoal treatments over 3 h postdosing may have been more appropriate.(20)
An alternative interpretation of the salmeterol pharmacokinetic data, without the need for an oral absorption component, can be considered. It was noted in the NGI test that DPI-A delivered more than twice as many particles of each drug in the range of 3–5 μm as did DPI-C (Fig. 2). The observed pharmacokinetics could be explained by a more rapid absorption of smaller particles (0.5–1 μm) from the alveolar spaces with DPI-C, and a slower and continued absorption of larger particles (3–5 μm) from the bronchial spaces with DPI-A.(6) However, one would also have to discount the large differences between products observed in the NGI apparatus in throat deposition and accept the supposition that the single charcoal pretreatment was able to completely prevent the oral absorption of swallowed drug. An additional clinical study with a more extensive charcoal block treatment is needed to test this hypothesis. It is our opinion that the salmeterol pharmacokinetics are more likely explained by a combination of pulmonary and oral absorption mechanisms.
Safety
Three patients reported adverse events during the study; none of these adverse events were related to study treatments. One adverse event (corneal abrasion) was reported during the screening phase, before any drug was administered. Two adverse events (caffeine-withdrawal headache and general headache) were each reported once, following the first time that study drugs were administered. Both adverse events were moderate in intensity and were treated with oral acetaminophen. The events resolved the same day. No other concomitant medication, including rescue medication, was administered during the study. No new adverse events were reported by any patient at the 1-month phone call.
Matching the total fine particle mass ≤3 μm for the products met the objective of assuring that DPI-C could be studied safely. Matching a total particle mass ≤5 μm, often considered the respirable mass, for the products was not utilized as this approach would have required a greater amount of each medication from DPI-C to be administered, particularly in the <3-μm region that we considered to be most critical for systemic absorption. For the first study in humans with DPI-C, we chose the more conservative route.
Evaluation of safety during this first-in-man study was limited to observation of adverse events. However, greater systemic exposure from fluticasone (both AUC and Cmax) and salmeterol (Cmax) are indicative of potential for greater systemic effects (adrenal suppression by fluticasone, and heart rate due to salmeterol) from DPI-C. Interpretation of comparable safety is further limited due the possible underestimate of the salmeterol Cmax because blood sampling initiated at 5 min. It is possible that with earlier blood sampling, higher salmeterol concentrations may have been observed.(21)
Conclusions
The aim of the reverse cyclone technology is to preferentially deliver small drug particles from a DPI that are potentially respirable while retaining larger lactose particles within the device. The lactose/drug clusters are exposed to the reverse cyclone action and deagglomerated prior to release to the patient. A simple cyclone mechanism in an experimental dry powder inhaler has been reported to reduce the mouth–throat deposition and to increase the pulmonary deposition of budesonide and ciprofloxacin in human mouth–throat casts and cascade impactor tests.(22) In the present study, DPI-C produced plasma profiles for both fluticasone and salmeterol that suggested a reverse cyclone technology in a commercial-ready device can result in a reduced mouth–throat deposition, and an increased pulmonary deposition. DPI-C was able to achieve comparable in vivo results for FEV1 while delivering only one-third the dose of DPI-A.
DPI-C was not expected to be bioequivalent to DPI-A. Our approach of matching the particle mass ≤3 μm for the products met the objective of assuring that DPI-C could be studied safely. If bioequivalence was an objective, matching of the individual cups (or perhaps groupings) of the NGI, as recommended by regulatory authorities, would have been utilized.(23)
Our results also underscore the difficulty in relying on pulmonary function to compare inhalation products intended to treat asthmatic patients. The well-established shallow dose response of pulmonary function indicates that equivalent local efficacy as measured by pulmonary function may be obtained with a range of products, but as the present study indicates, the systemic component of these products, which contributes to safety, may be widely different.(23) Therefore, we have limited our conclusions to the doses, patient population, and the generally recognized limited sensitivity of the bronchodilation model used in this study.
In this population of patients with mild-to-moderate asthma, it can be concluded that both DPI-C and DPI-A had a similar effect on the improvement in FEV1 and showed a comparable lack of adverse events. Pharmacokinetic results suggested that DPI-C deposited more drug in the lungs and less drug in the mouth and throat than did DPI-A. These preliminary results need to be confirmed in larger studies.
Footnotes
Acknowledgments
An abstract of this study was presented at Respiratory Drug Delivery Europe 2011, Berlin GE, May 3–6, 2011. We gratefully acknowledge members of the 3M staff who contributed to device preparation: D. Riley, J. Clay, N. Sivewright, A. Dasani, S. Parker, P. Cocks, S. Halling, E. Ross, A. Slowey, P. Bainbridge, S. Dexter, K. Hunt, A. Stuart, A. Henry, D. Hodson, G. Hill, D. Pearson, S. Dee, G. Adlam, M. Lightfoot, N.H. DO, C. Stevenson, R. Sitz, M. Emery, J. Kurle, J. Miller, J. Beaurline, and S. Stein; members of Sylvanna staff who performed the study: S. Syring, J. Woodworth, E. Hernandez, L. Santana, and R. Magee: members of PPD staff who monitored the study: S. Worah, S. Fields, and K. Griffith; members of PPD Bioananlytical who quantitated the analytes in plasma: Jim Swanson, L. Pendracine, and A. Dorzweiler.
Author Disclosure Statement
L. Harrison, C. Novak and M. Needham are employees of 3M Company; L. Harrison and C. Novak own stock in the company. No conflicts with this study exist for P. Ratner.