
Abstract
Abstract
Background:
Nanoparticles (NPs) produced by nanotechnology processes have taken the field of medicine by storm. Concerns about safety of these NPs in humans, however, have recently been raised. Although studies of NP toxicity have focused on lung disease the mechanistic link between NP exposure and lung injury remained unclear. This is primarily due to a lack of availability of appropriate airway disease models and sophisticated microscopic techniques to study nano-sized particulate delivery and resulting responses.
Methods:
Air–liquid interface (ALI) cultures of non-cystic fibrosis (CF) and CF airway epithelial cells were exposed to the FITC-labeled NPs using a PennCentury microsprayer™. Uptake of NPs was assessed by FACS. Laser scanning microscopy (LSM) was performed and the images were analyzed by an advanced imaging software to study particle deposition and uptake.
Results:
Flow cytometry data revealed that CF cells accumulated increased amounts of NPs. The increased NP uptake could be attributed to the reduced CF transmembrane conductance regulator (CFTR) function as a similar increased retention/uptake was observed in cells whose CFTR expression was downregulated by antisense oligonucleotide. NPs alone did not induce pro-inflammatory cytokine release or cell death. The cell culture system was sensitive to ozone but exposure to the uncoated synthetic NPs used in this study, did not cause any synergistic or suppressive effects. LSM imaging and subsequent image restoration further indicated particle uptake and intracellular localization. Exposure to ozone increased nuclear uptake in both non-CF and CF cells.
Conclusion:
Our findings demonstrate the uptake of NPs using ALI cultures of non-CF and CF airway epithelial cells. The NPs used here were useful in demonstrating uptake by airway epithelial cells without causing adverse effects in presence or absence of ozone. However, to totally exclude toxic effects, chronic studies under in vivo conditions using coated particulates are required.
Introduction
NPs have a wide range of applications, and nanotechnology is promising in many fields of medical applications, including cancer treatment, drug targeting, and contrast agents.(4) Although a large number of very attractive applications open up for the clinics, one needs to be very careful in allowing new nanomaterials for human use because of their potential toxicity.(5,6) Adequate knowledge of their particokinetics(7) and toxicity is required to counterbalance the vigorous research on new nanomaterials that are potentially useful in medicine. The numerous nanocarriers used to transport and release therapeutic molecules to the target tissues should be treated as additives, with potential side effects of themselves or by virtue of their solubility/potential dissolution in biological media or aggregation inside the body. This would not only improve the existing nanomaterials but also bring forward novel improved nanoparticles with reduced adverse effects.
Not only has the use of nanomaterial increased, their atmospheric presence has elevated as well. With every breath we unintentionally inhale millions of nano-sized particulates, mainly originated from combustion processes but more and more also some of them deliberately as engineered nanoparticles, for example, in the form of aerosolized drugs for treatment of respiratory ailments. Airborne particulate matter (PM) increases morbidity and mortality from cardiopulmonary diseases with increasing toxicity as PM size decreases.(8) Further, there is an increased precedence of air pollution-induced chronic airway disease exacerbation such as that of asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF). CF is a fatal inherited disease caused by the mutation in the CF transmembrane conductance regulator (CFTR) gene. Cardiopulmonary failure accounts for 95% of CF-related deaths.(9–11) Exposure to airborne PM has been shown to be associated with increased pulmonary exacerbation and progressive decline in lung function in CF.(9–11) Although there is immense literature about the interaction of NPs with normal airways and airway epithelial cells, investigations involving diseased airways are unknown. There is also a gap in understanding of as to how atmospheric pollutants such as ozone modify the particulates by itself or the interaction with cells of the airways and the response of the airways. In this study we have used engineered NPs, that is, polystyrene NPs to investigate their deposition and interaction with CF airway epithelial cells in absence or presence of ozone and compared them with those of non-CF.
Materials and Methods
Airway epithelial cell culture
Normal and CF cell lines
CF41o-, CF45o-, and a wild-type (healthy/non-CF) airway epithelial cell line, 16HBEo-, were provided by Prof. D. Gruenert (California Pacific Medical Center Research Institute, University of California at San Francisco). They were cultured in Eagle's minimal essential medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS), L-glutamine, and penicillin/streptomycin at 37°C under 5% CO2. 16HBEo- cells with stable expression of sense (16HBE-S or S-1) and antisense CFTR (16HBE-AS or AS-3) oligonucleotides were cultured as described previously.(12) Polarized cultures growing on fibronectin-collagen coated Snapwells™ (0.4-μM pore size, Corning, Corning, NY). These Snapwells™ are inserts that are suspended on a frame that hangs in a six-well plate.
Primary healthy (non-CF) and CF airway epithelial cells
Primary human bronchial epithelial cell harvest and culture was performed using established procedures previously described in detail(13) under National Jewish institutional review board (NJIRB)-approved protocols. Primary epithelial cells (passage 2; 5×105 cells) were seeded onto 12-mm diameter type VI collagen (Sigma, St. Louis, MO)-coated Snapwells™ (0.4-μM pore size, Corning, Corning, NY). Following confluence on days 4–5 the cultures were maintained at an air–liquid interface (ALI). All experiments comparing primary non-CF and CF cells were performed with ALI cultures grown simultaneously and matched for passage number, number of cells plated, and days in culture. Undifferentiated cultures of primary airway epithelial cells obtained from three non-CF (normal) and three CF subjects were used (detailed description in Ahmad et al.(14)).
Trans-epithelial electrical resistance (TEER) was measured as described previously.(15,16) For TEER measurement medium was added to the upper chamber 30 min before TEER was measured and again removed after the measurements. Addition of media apically provides solution on the upper surface to dip one arm of the chopstick electrode and is necessary for TEER measurement.
Particle exposure
After 7 days in culture the cells were exposed to air for 24 h, and then particles (aerosolized) were sprayed on the apical surface of the cultures with a Micro Sprayer model IA-1C with a 10" long 0.64-mm tube (PennCentury™, Philadelphia, PA).(16, 17) Particle suspension was prepared as follows: fluoresbrite microspheres, plain yellow green with a diameter of 0.05 μm, were obtained from Polyscience (Brunschwig, Basel, Switzerland). The particle dimensions were confirmed using a Zetasizer (nanoseries ZEN 3600, Malvern Instruments Ltd, Worcestershire, UK) to be of 40.4±3.1 (mean value±SEM) nm. We also characterized the charge of the NPs as −35.2 mV as it was not disclosed by the manufacturer but was anticipated as they are solubilized at a step in SDS, which is subsequently removed during manufacture. The details of NP size determination and associated discrepancies have been previously described.(18) Particles were diluted to a concentration of 6.5×105 particles/μL in media (MEM alpha for cell lines and ALI media, as defined in Ahmad et al.,(14) for primary cells) and sonicated (1 mL of the NP dispersion was sonicated with a Sonicator Q55 50-Watt, 20-kHz sonicator, 30% intensity; Qsonica, LLC, Newtown, CT) for 10 sec three times. For adequate particle distribution on the sample, 25 μL (1.5×107particles) of particle suspension were sprayed on each air-exposed culture (>3.6×106 particles/cm2 or 0.1 μg/cm2). After particle exposure the cell cultures were incubated for further 24 h, then exposed to ozone [the particle exposure was done first and the same cell cultures (snapwells) were exposed to ozone], washed with PBS, and fixed.
Flow cytometry
The day after particle exposure cells were harvested by trypsinization and suspended in 1.0 mL Hank's Balanced Salt Solution (HBSS) containing 2.0 μg/mL propidium iodide. The samples were then put on ice. Analysis was done using a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA).
Ozone exposures
Exposure of airway epithelial cells to ozone at precise levels was carried out in a computer-controlled in vitro exposure chamber.(15) Briefly, the exposure system consisted of four identical exposure systems maintained in a single temperature-controlled (37°C) environmental chamber (Forma Scientific, Marietta, OH) controlled by a single desktop computer. One of these four systems was always used for an air control (0 ppb ozone) while the other three could be used for exposure of cells to different ozone concentrations. Ozone was produced by bubbling medical-grade compressed oxygen through a coldspark corona discharge ozone generator (Model OZ2SS-SS, Ozotech, Yreka, CA). The air/CO2 mixture was directed into the environmental chamber where it was warmed and humidified by bubbling through a glass water bath containing 1.5 L of water thermostatically maintained at 37°C. Upon exiting the water bath, the warm air/CO2 was mixed with the ozone/oxygen stream and then passed to a glass exposure chamber containing the cells to be exposed. Cells growing on snapwells suspended in a six-well plate with 100 μL media on top were gently rocked inside the chamber (16 sec, tilt time four times a minute) so as to expose one side of culture well at a time directly to ozone. Gas flow through the chambers for these experiments was maintained at 5 L/min. Humidity of the chambers was 95±5%.
Interleukin (IL)-8 assay
At the end of exposure additional 200 μL media was added apically. Supernatant media was collected after 4 h and analyzed for IL-8 by ELISA (ElisaTech, Denver, CO) as described before.(19)
Cell labeling and fixation
Cell cultures were fixed and stained as previously described. 20 Antibodies were diluted in PBS as follows: nucleic acid stain DAPI (Molecular Probes, Juro Supply GmbH, Lucerne, Switzerland) and rhodamine phalloidin (that stains F-actin) 1:100 (Molecular Probes).
Laser scanning microscopy and image restoration
A Zeiss LSM 510 Meta with an inverted Zeiss microscope (Axiovert 200M; lasers: HeNe 543 nm, and Ar 488 nm; Carl Zeiss AG, Feldbach, Switzerland) was used. Image processing and visualization was performed using IMARIS (Bitplane AG, Zurich, Switzerland), a three-dimensional multichannel image processing software for confocal microscopic images.(16,20) To visualize the labeled NPs inside the epithelium, a rendering mode was used, which shows the maximum intensity projection (i.e., the maximum intensity of all layers along the viewing direction) of the recorded three-dimensional stack. To illustrate the “luminal” surface, a shadow projection was applied from different observation angles. For the visualization of three-dimensional data sets, particularly for the localization of particles inside the cells, the surpass module from IMARIS was used, which provides extended functions: the volume rendering, which displays the volume of the entire data set, or the IsoSurface visualization, which is a computer-generated representation of a specific grey value range in the data set. It creates an artificial solid object to visualize the range of interest of a volume object.
Statistical analysis
All statistical calculations were performed with JMP and SAS software (SAS Institute, Cary, NC). Means were compared either by two-tailed t-test for comparison between two groups or one-way analysis of variance (ANOVA) followed by the Tukey-Kramer test for multiple comparisons for analyses involving three or more groups. A p-value of <0.05 was considered significant.
Results
Particle exposure and uptake
The particle exposure was performed at the ALI of cultures of non-CF and CF cell lines 16HBEo-, CF41o-, and CF45o-, respectively. As described in the Methods section about 3.6×106 particles/cm2 were sprayed. We recovered about 55±9% (mean±SEM) of fluorescence from the exposed cells indicating a deposition efficiency of approximately 55%. Using this approach we have previously demonstrated using particles with a diameter of 1 mm a deposition efficiency of about 60% and within 24 h about 40% particle uptake by the cells.(16) The fluorescent NPs served as an excellent tool for their quantitation in cells. In addition, propidium iodide was used as marker for cell death. Increased amounts of NPs were retained in the airway epithelial cells. CF cells showed increased particulate-conjugated FITC fluorescence, especially CF45o- cells exhibited a significantly greater mean fluorescence intensity (MFI) compared to the non-CF 16HBEo- cells (Fig. 1A). Exposure to NPs did not cause cytotoxicity as indicated by a minimal propidium iodide uptake (Fig. 1B).
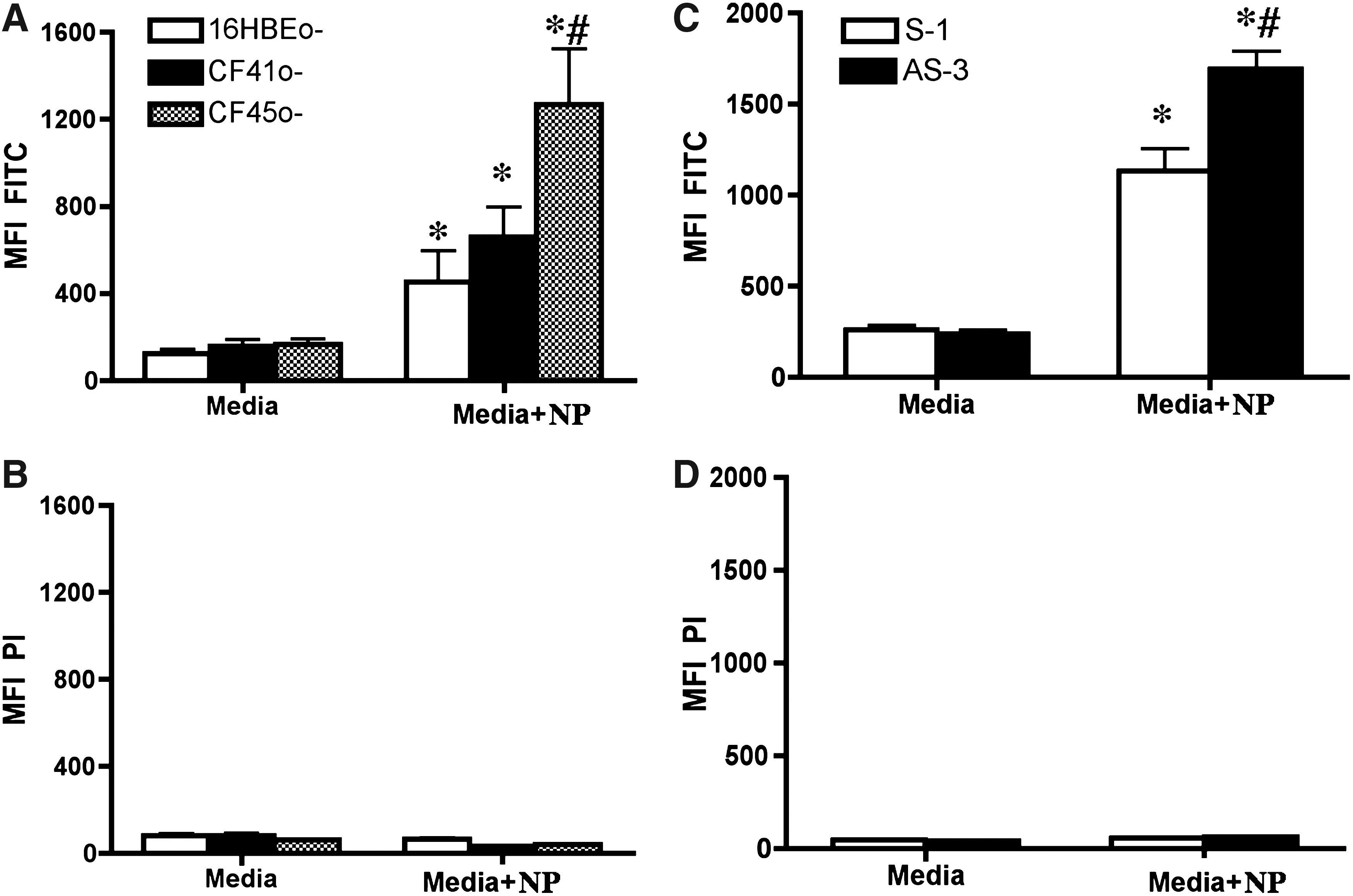
Nanoparticle (NP) uptake by non-CF and CF airway epithelial cells. Non-CF (16HBE0-) and CF (CF41o- and CF45o-) cells were cultured on collagen/fibronectin-coated inserts for 7 days. On the seventh day the apical media was removed and the ALI culture was exposed to the NPs on the eighth day as described in the Methods section. After 24 h cell layers were washed with warm PBS and harvested. Propidium iodide (2.0 μg/mL) was added to assess cell death and the fluorescence (mean fluorescence intensity, MFI) was measured using a flow cytometer (A, B). Similarly non-CF (CFTR sense oligonucleotide expressing 16HBE cells, S-1) and CF (CFTR antisense oligonucleotide expressing 16HBE cells, AS-3) cells were cultured and exposed to the NPs and particle fluorescence (
We used cells expressing antisense CFTR oligonucleotides to evaluate if CFTR activity plays a role in particulate retention. Our results demonstrated that cells with decreased CFTR activity, due to CFTR knockdown, had increased retention of NPs (Fig. 1C). Exposure to NPs was not associated with cytotoxicity in these cells as well (Fig. 1D).
Effect of NPs on the TEER
To further evaluate the cellular integrity TEER was measured at various time intervals (0–6 h) after NP exposure (Fig. 2). No significant change was observed in the TEER upon NP exposure in the non-CF (16HBE) and CF (CF41o-) cells (465±40 ohms cm2, mean±SEM in 16HBEo- and 324±38 ohms cm2 in CF41o- cell before exposure and 444±34 ohms cm2 in 16HBEo- and 345±45 ohms cm2 in CF41o- cells after 6 h of exposure.
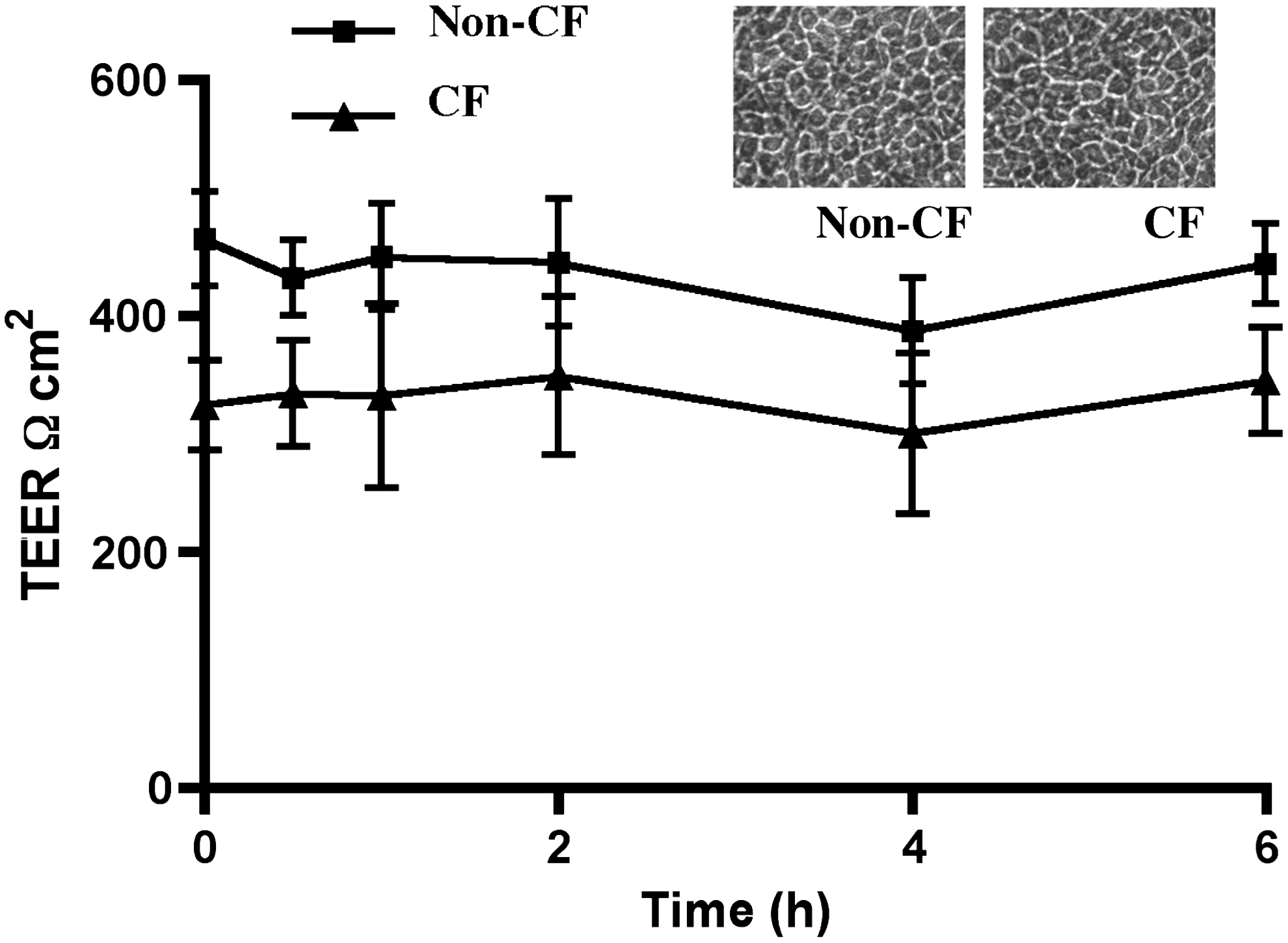
Effect of NP exposure on the airway epithelial cell resistance. Non-CF (16HBE) and CF (CF41o-) cells were cultured and treated as described in the legend to Figure 1. Transepithelial electrical resistance (TEER) was measured at various time intervals as described in the Methods section. The inset shows the morphology of non-CF and CF cells at exposure.
Effect of NPs on ozone-induced pro-inflammatory cytokine release and cell death
We have previously demonstrated ozone-induced apoptotic cell death and pro-inflammatory cytokine release in airway epithelial cells.(15,19) Our results have previously (unpublished) demonstrated that polarized cultures of CF airway epithelial cell lines have enhanced cell death upon exposure to higher concentration of ozone (500 ppb). Here we investigated the effect of particulate exposure on interleukin (IL)-8 release and cell death in non-CF and CF airway epithelial cells and whether these are modulated by ozone. Our studies demonstrated increased IL-8 release and enhanced apoptotic cell death upon ozone exposure in non-CF (16HBEo-) airway epithelial cells (Fig. 3A and B). CF (CF45o-) airway epithelial cells exhibited further enhanced IL-8 release and apoptotic cell death compared to non-CF airway epithelial cells. However, particulate exposure prior to ozone exposure did not further exacerbate the ozone toxicity in non-CF or CF cells (Fig. 3A and B).
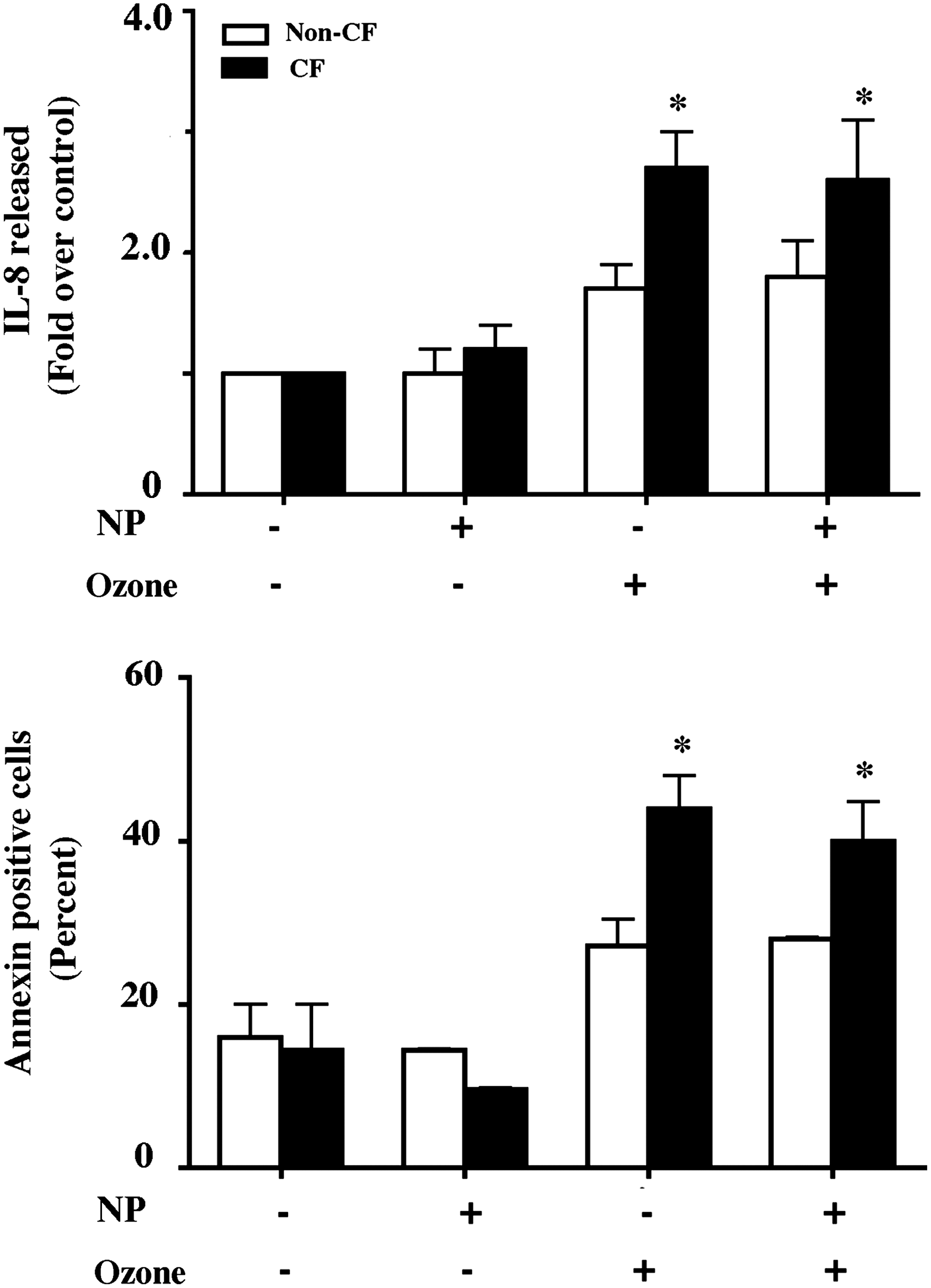
Effect of NP on ozone-induced airway epithelial cell cytokine release and death. For IL-8 assay non-CF (16HBE) and CF (CF41o-) cells were cultured and treated as described in the legend to Figure 1 and exposed to 500 ppb ozone for 8 h. At the end of exposure additional media was added apically and at 4 h apical media was collected for IL-8 assay and cells were lysed for protein assay (
Localization of NPs by laser scanning microscopy (LSM)
We used primary non-CF and CF airway epithelial cells to study their interaction with NPs. These cells form tighter junctions and have the ability to differentiate when provided appropriate conditions.(14) Fluorescent NP localization in primary airway epithelial cells was performed by immunostaining methods in combination with LSM and digital image restoration (Fig. 4). After the NP fluorescence was detected, reconstruction of confocal data was performed to obtain three-dimensional images as described previously.(20) NPs were observed inside the cells that were also stained for cytoskeletal F-actin (Fig. 4a–d).
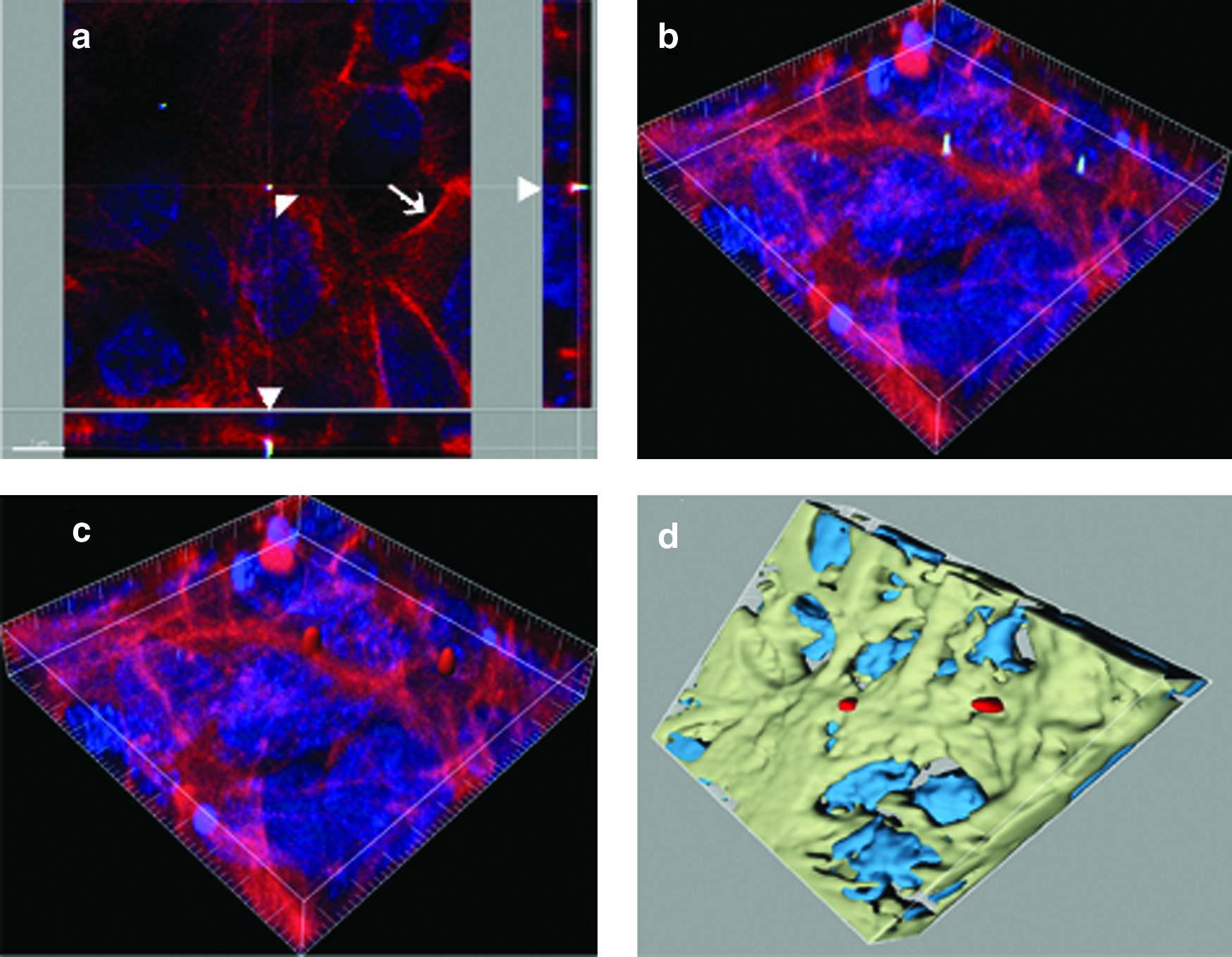
Laser scanning microscopy (LSM) and image restoration for the visualization of NP inside airway epithelial cells. Representative LSM images of primary airway epithelial cells grown on collagen-coated inserts (
NP localization in primary non-CF and CF cells and effect of ozone
Using the approach described above we analyzed particle localization in air and ozone exposed air–liquid interface cultures of primary non-CF and CF airway epithelial cells. Very few NPs localized at the surface and most of them appeared inside the cells (Fig. 5). Three images from each sample were scanned and analyzed to locate the particle and the results are shown in Figure 6. All the cells within each image field were analyzed. The results further demonstrated an intracellular localization of the NPs in the air exposed ALI cultures of non-CF and CF cells. Exposure to ozone caused an increase in intranuclear delivery (Fig. 5, e–h and m–p and Fig. 6) of the PM with an equal distribution in both non-CF and CF cells.

Localization of NPs in non-CF and CF airway epithelial cells: cells were cultured for 7 d on the inserts and treated with NPs and exposed to ozone (0 ppb or 200 ppb for 8 h), fixed, and stained for F-Actin as described in the text. For the localization of particles inside the cells, the surpass module from IMARIS was used as shown in Figure 4. Red color was used for the NPs (white arrowhead), green represents the F-Actin in the cytosol (white arrow), and blue the DAPI stained nucleus (empty white arrowhead) shown in
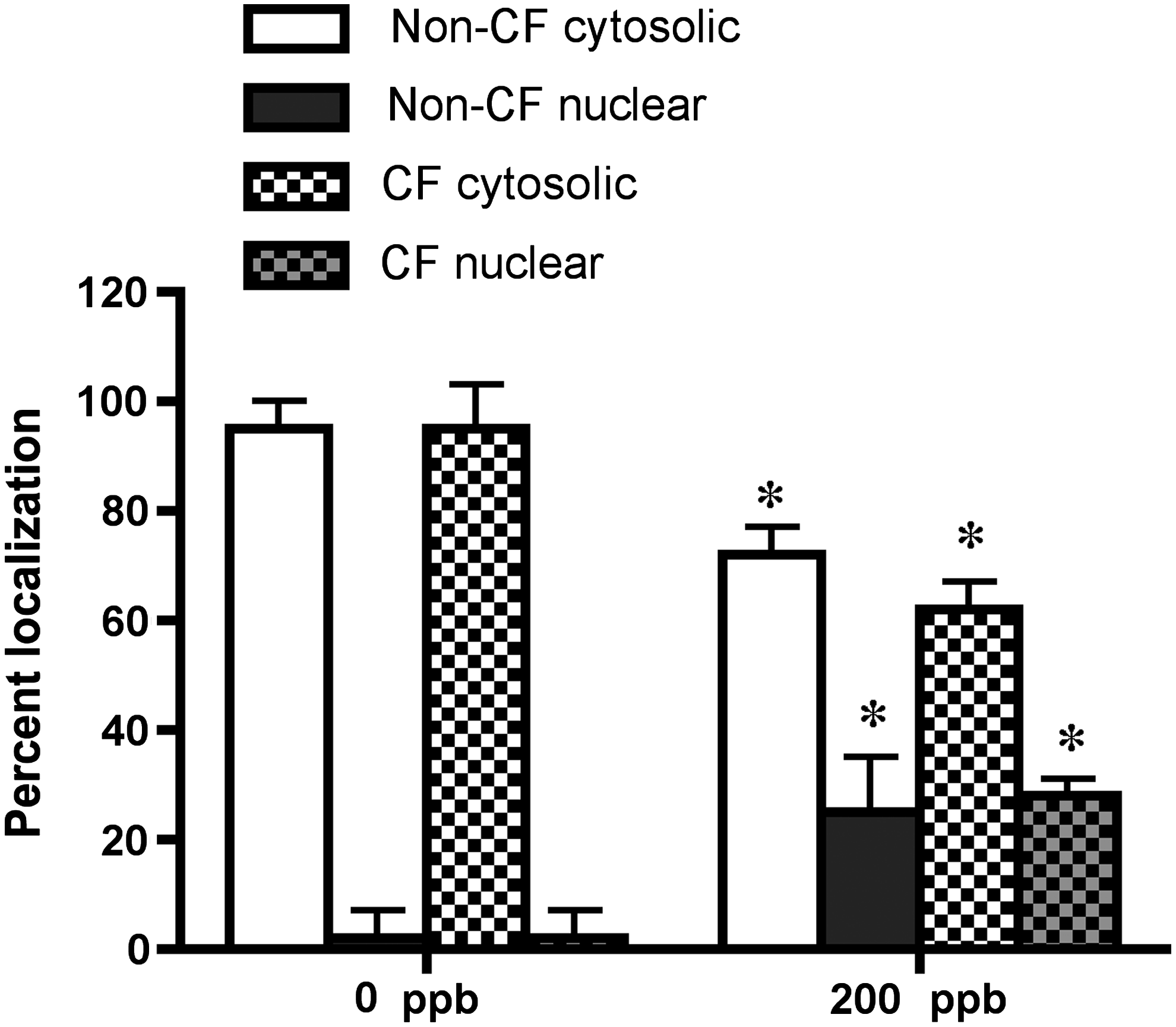
Visual quantitation of NP partitioning inside cells and nuclear compartment in non-CF and CF cells. Primary non-CF and CF airway epithelial cells were cultured for 7 days on the inserts and treated with NPs and exposed to ozone (0 ppb or 200 ppb for 8 h), fixed, and stained for F-Actin as described in the text. Values are mean±SEM (n=3 per group per donor; cells from three individual donors each from non-CF and CF subjects were used). *Indicates significant difference (p≤0.05) from 0 ppb-exposed cells.
Discussion
Due to its large surface area, the lung is the major site of interaction with inhaled NPs.(21,22) Once a certain type of NP can be identified unambiguously using microscopic methods it is desirable to quantify the particle distribution within a cell, an organ, or the whole organism. We have previously demonstrated using TEM qualitative and quantitative analyses of particle-related structural changes of the respiratory tract, revealed the localization of nanoparticles within tissues and cells and investigated the 3D nature of NP-lung interactions.(23,24) A triple cell coculture system, composed of epithelial cells, macrophages, and dendritic cells, was developed that simulates the most important barrier functions of the airway epithelium.(25) Using this system the intracellular localization of titanium dioxide nanoparticles was analyzed by energy filtering transmission electron microscopy.(26) Titanium dioxide NPs were detected as single particles without surrounding membranes and in membrane-bound agglomerates. The triple cell coculture incubated with titanium dioxide particles also showed an elevated production of reactive oxygen species but no increase of the release of tumor necrosis factor alpha. The interplay of different lung cell types seems to substantially modulate the oxidative stress and the inflammatory responses after NP exposure.(27) Our recent studies have also shown that fluorescent-magnetic hybrid NP-induce increased pro-inflammatory responses in airway epithelial cell cultures; on the other hand, gold NPs did not cause adverse effects.(28,29) These studies suggest that particulate characteristics and cell culture models may have substantial impact on the cellular responses.
The cellular responses may further be modified by disease states of the donors and the environmental stresses and need to be evaluated.(30,31) With this aim we designed this study to investigate cellular deposition and delivery in normal and diseased airway epithelial cell culture models. In the current study NPs were exposed to cells at ALI that resemble in vivo exposure conditions of the lungs more realistically than under submerged conditions and allows a controlled deposition of aerosolized NPs.(32) The use of 3.6×106 particles/cm2 which corresponds to 0.1 mg/cm2 is considered to be in the realistic range as recommended by Occupational Safety and Health Administration standard.(32) However, in the present study the administration of the polystyrene NPs was done with the PennCentury microsprayer™, and with this system it is only possible to deliver the dose within a very short time, that is, few seconds. This does not correspond to the in vivo situation where an aerosol is inhaled over time. Several air–liquid exposure systems are meanwhile described in the literature such as CULTEX or the ALICE, and with those devices it is possible to apply an aerosol over time, that is, mimicking again a more realistic situation.(33–35) For future studies such more sophisticated systems are planned to be applied.
We also evaluated ozone, an important environmental factor, for its potential effects on modulating the NP delivery and response of airway epithelial cells. We demonstrate that ALI cultures of non-CF and CF airway epithelial cells accumulate increased quantities of synthetic NPs. Flow cytometry data revealed increased fluorescence in the CF cell lines, suggesting enhanced particle uptake. This could be due to CFTR dysfunction that may contribute to decreased vesicular exocytosis. Inhibition of exocytosis and accumulation of antibiotics has been shown previously in the epithelium of nasal polyps in CF patients.(36) The TEER of the tight ALI culture of non-CF and CF cells was not modulated upon interaction with NPs, indicating that the particles do not cause any changes in the properties of tight junctions. The TEER values in our cell cultures were close to what has been previously demonstrated by us and others under similar duration and conditions.(15,25,37) Preservation of tight junctions upon particle exposure has been also shown in our previous studies.(20)
Imaging of the NPs using LSM in the airway epithelial cells did not reveal any particle within the junctions but only attached to the surface of the cells or in the cells. Ozone-induced nuclear translocation of NPs is a novel finding and has not been demonstrated before. Specific peptide mediated targeted delivery of gold NPs has been shown before.(38,39) Moreover, there are evidences that support nuclear accumulation as an methodological artifact.(40) Our studies, however, carefully incorporate normal and diseased air exposed controls that exclude such nonspecific deposition. The mechanism of the nuclear uptake is not clear, given that size of nuclear pore is smaller than NPs used in this study. Ozone exposure may contribute to a transient nuclear pore modulation to cause uptake of the particle. A nonspecific transiently increased nuclear pore size, allowing entry of larger particles, upon inhibition of protein synthesis has been demonstrated before.(41) Therefore, particulate entry in nucleus may be a nonspecific uptake. The nuclear pore does disassemble and quickly reassemble, potentially capturing particulates in the process of cell division but cell cultures growing on inserts normally do not divide. Role of nucleolin, a protein of the nucleolus in trafficking of nanoparticles and its modification by steroids has recently been demonstrated.(42) Nuclear entry of polylysine nanoparticles (73 nm hydrodynamic diameter) was also observed in chochlear cells.(42,43) Further confirmatory methods may be required to study this phenomenon.
Development of aerosolized drug delivery for various airway diseases including CF has improved considerably.(44,45) Novel, functionalized NPs and coatings have already shown promise.(46) Further, modified magnetofection techniques to enable penetration of physical barriers such as mucus show improved benefit for delivering therapeutic genes to treat CF.(47) However, studies on toxicity, to evaluate safety of such materials, are generally focused on particles greater than 1 μm. Moreover, studies evaluating the impact of environmental factors on particle deposition and toxicity are absent. Although our results demonstrate that the synthetic NPs used in this study do not modulate ozone-induced pro-inflammatory response and cytotoxicity of non-CF and CF airway epithelial cells it is important to reevaluate the effect with each NP of interest.
In conclusion, this study demonstrates detection and localization of NPs in normal and CF cells cultured at ALI by flow cytometry and by LSM. Using advanced image reconstruction techniques we provide the ability to locate NPs in the cell nucleus when ozone was applied. We also demonstrate that NP exposure does not modify the (pro-) inflammatory response of the airway epithelial cells to ozone. Ozone exposure would, in turn, influence responses to particulates and would be interesting to evaluate. Further studies involving evaluation of functionalized or environmental NPs in normal and diseased organs such as lung and the effect of atmospheric factors such as ozone using further improved exposure systems (that achieve aerosol delivery over prolonged periods that actually occur) are required to carefully rule out potential adverse effects.
Footnotes
Acknowledgments
This work was supported by R01-ES014448 from the National Institute of Environmental Health Sciences (NIEHS), NIH, and by the Max and Yetta Karasik Family Foundation (CWW). S.A. is supported by K12 KL2RR025779 from the National Center for Research Resources (NCRR, NIH). A.A. is supported by the Scientist Development Grant award (0830418N) from the American Heart Association. B.R.R. and D.R. are supported by the Lung league Bern, Switzerland, the German Research Foundation (DFG SPP 1313) and the Swiss National Foundation (No. 3100A0_118420). The authors are grateful to Dr. Scott Randell, University of North Carolina (UNC), North Carolina, for providing primary CF airway epithelial cells and Dr. Pamela Davis, Case Western Reserve University Cleveland, OH, for the CFTR-S and CFTR-AS expressing 16HBEo- cells.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.