
Abstract
Abstract
Two separate international standards, ISO 20072:2009 and ISO 27427:2010, have recently been published that relate to the development and performance testing of oral inhaled products (OIPs). The scope of ISO 20072 encompasses all OIP forms except nebulizing systems, whereas ISO 27427 was developed specifically for this class of OIP. Compliance with these standards will likely be necessary for manufacturers seeking approval to market inhaler devices in the European Union (EU). Their adoption in the United States may take a considerable time, but the FDA has expressed support in general terms for the ISO process. Key aspects of both standards that are very different in style and content are identified and discussed from the perspective of a potential user. In the approach adopted by ISO 20072, a formalized risk assessment is undertaken as a key part of design verification, in order to develop the Device Functionality Profile (DFP) of the device. The DFP is subsequently verified by the System Verification Test (SVT), in which pharmacopeial test methods are used to evaluate in vitro performance of the device with a chosen drug product in a statistically robust manner. On the other hand, ISO 27427 adopts a more prescriptive approach that involves performance verification of the finished nebulizing system using 1% w/v salbutamol as the test formulation. Although ISO 27427 is currently undergoing revision, at present it is unclear whether the changes that are made will significantly alter its fundamentally different approach to device performance verification. A strong case can be made for a single OIP-wide ISO standard, based on the principles developed in ISO 20072 and that makes use of the well-understood and validated in vitro test procedures that are available or will shortly be available in the case of nebulizing systems, in the United States and European pharmacopeias.
Introduction
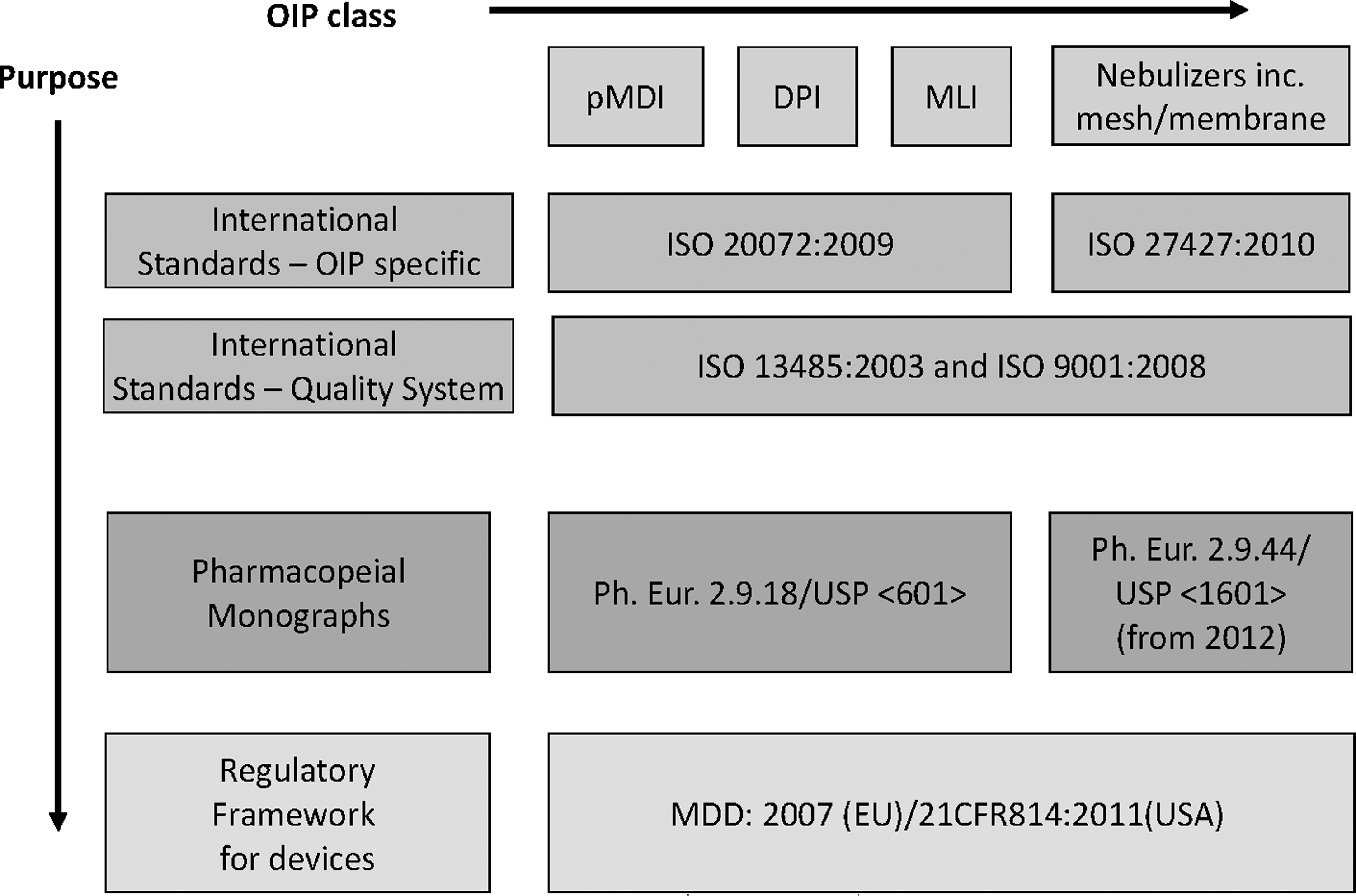
Relationships between the regulatory framework, ISO standards, and the pharmacopeial monographs in the United States and EU for the development of the device aspect of OIPs.
The primary purpose of the regulatory and standards framework is to provide direction to the manufacturer for the safe and effective development of a medical device. However, the documents mentioned above do not go further to indicate how the device and formulation (drug product) interact to conform within the intended specifications set by the manufacturer for the final product that is marketed. Historically, in Europe, this interaction had been dealt with by the drug product manufacturer at a late stage in the development process when submitting the dossier of performance data for the formulation-in-device combination to the regulatory body governing the intended market. If an independent check of device performance with the formulation in question had not been undertaken earlier in the drug product development process, the outcome could be both unpredictable and potentially expensive to rectify deficiencies. As a consequence, in the early 2000s, it was seen by many stakeholders in the device development process, as advantageous to have an internationally applicable standard that combines the appropriate guidelines, directives, and national standards (where applicable). Almost from the outset, it was foreseen that such a standard would ideally adopt a user-guide approach to test the drug product–device combination early in product development process, preferably at the design verification stage for the device component.
The consensus within industry-wide bodies such as the European Pharmaceutical Aerosol Group (EPAG), who became involved early on in the public commentary to the process, was that if an ISO standard was to be created to provide guidance to the device design verification process, it should encompass all types of oral inhalers within its scope. However, for various reasons explained in more detail below, a division into two separate standards ultimately took place.
The Development Process for the Two ISO Inhaler Standards
OIPs can be conveniently divided into three major classes having substantially different medication delivery characteristics, involving more or less active interaction by the patient. These are:
1. dry powder inhalers (DPI), 2. pressurized metered-dose inhalers (MDI) together with nonpressurized metered liquid inhalers (MLI), 3. nebulizing systems.
Today, all inhalers, with the possible exception of MDIs, can readily incorporate electrically operated components for a variety of purposes, most usually to provide a source of power independent of the user, but also for certain devices to provide guidance to the patient or caregiver during administration of the medication. In the particular case of nebulizing systems not operating from a fixed pressurized air or oxygen supply as is available in hospital use, electrical power can be provided to operate the device either by a hand-held, portable battery, or by a fixed mains-powered compressor. The incorporation of such components significantly complicates the design process beyond that associated with devices that comprise only mechanical parts.
A further challenge to the device developer in the case of the MDI class of OIPs has been the change in formulation technology associated with the transition from chlorofluorocarbon to hydrofluoroalkane propellants. This transition was undoubtedly more of an issue for the drug product developer, but considerations such as metering valve design(8) proved to be as much an exercise for the developer of the MDI canister as for the formulators. In addition, there was the increasing pressure from regulators, particularly in the United States, to incorporate aids that inform the user of the number of actuations of medication remaining.(9)
Until recently, the device part of both MDI and DPI inhaler groups had not had standardized guidance that could be used to evaluate systematically in vitro performance of critical aspects of the drug–device combination as part of design verification, because the compendial test methods are largely focused on product quality control (i.e., batch release testing).(10) The components used to deliver the medication were not tested as devices separately from the drug product, partly due to the fact that these OIPs were seen as integral packages associated with a specific drug product (i.e., the canister-valve system forming the drug product containment closure system in the case of MDIs). The need to look at the device performance in isolation from the drug formulation was therefore not evident. In contrast, for some time nebulizer performance has been assessed as a device independently from a particular drug product during the design verification process, initially in the United Kingdom by means of the now obsolete British Standard document BS7711-3:(11) 1994. In the late 1990s, a revision to BS7711 was undertaken within the EU as part of the mandate of the European Committee for Standardization (CEN) to supersede national standards. The aim of the new standard published initially in 2001 specifically covering nebulizers,(12) was to identify and implement a more appropriate assessment technology for this class of OIP, at the same time making this methodology available throughout the EU. The CEN nebulizer standard was supported by a clinical guideline developed by the European Respiratory Society(13) in an attempt to marry in vitro performance testing with methods that were deemed to be capable of providing clinicians with useful information to improve nebulizer therapies. However, the CEN standard demanded more elaborate work to be performed than what was needed for the predecessor BS7711-3 standard; this was mainly due to the assessment technology of the CEN standard that implemented both collection and assay of the aerosol. The laboratory-based testing procedures in BS7711-3 included the application of a laser diffraction method to size-analyze the aerosol produced by the nebulizer without an assay of the pharmaceutical content of the aerosol. From an industry perspective, the implementation of the CEN standard increased the knowledge of clinically valuable information of nebulizer in vitro performance at the cost of increased workload.
In the early 2000s Working Group 5 of ISO Technical Committee 84 “Devices for administration of medicinal products and intravascular catheters” (TC 84-WG5) of ISO identified from stakeholders a need for a standard for the development of hand-held inhalation devices that would include MDIs and associated add-on devices (spacers and valved holding chambers), DPIs and certain specialized MLIs that were not deemed to be nebulizing systems. ISO 20072:2009(14) was subsequently developed by Joint Working Group 5 between 2001 and 2008. Almost simultaneously Subcommittee 2 of ISO-TC121 “Anaesthetic and respiratory equipment” (TC121-SC2) initiated the development in parallel of a standard covering all types of nebulizing systems. ISO 27427:2009, revised in 2010,(15) covers jet, ultrasonic, and the newer vibrating mesh/membrane nebulizers. Its scope also encompasses nebulizers that operate either continuously or with breath enhancement/actuation. Although there was some dialogue between the two committees responsible for these inhaler-based standards, attempts to harmonize their approach and purpose failed to achieve this important goal. ISO 20072:2009 became a device design verification standard, while ISO 27427:2010 has remained a finished device performance standard, much like its predecessor, EN13544:2001.
ISO 20072:2009
The scope of this standard applies to: “the design, labeling, instructions for use and testing requirements for hand-held single- and multi-use aerosol drug delivery devices (ADDDs) intended to deliver a metered or pre-metered aerosolized medication to or by means of the human respiratory tract (including nasal, oral, tracheal, bronchial and alveolar sites).”
ISO 20072 was able to evolve from first principles, as there was no predecessor standard to take into account that could influence the development process. In essence, this meant that “state-of-the-art” procedures, methods, and technology could be included when developing inhalation devices. Its developers were able to recognize the broad variation in current device designs, and could develop a flexible approach in the descriptions of both general and specific requirements, as well as to the design of the testing regimen in order to cope with foreseeable developments in the near future. Early on, the working group decided in favor of making the process risk management based, in agreement with that described in ISO 14971:2007(16) (Medical Devices—application of risk management to medical devices), rather than resorting to a prescribed formula of requirements and associated normative testing. This more flexible approach is in harmony with current regulatory concepts such as the FDA interpretation of Quality-by-Design (QbD),(17) involving a comprehensive understanding of a process rather than the traditional prescriptive approach with fixed (and often limited) considerations, followed by testing to verify compliance. During the design verification process, the user therefore identifies the critical risk factors (analogous to critical quality attributes in the context of QbD) that may arise affecting the aerosol drug delivery device (ADDD) behavior in the hands of the eventual user (Fig. 2). The process then provides the option to eliminate or minimize their effect. Testing therefore becomes an integral part of checking that each risk factor has been appropriately managed through device development. It is important to note that this procedure is meant to be seen as being iterative in nature, because it is highly probable that several attempts will be needed to resolve the more intractable problems, ensuring that their impact in the hands of the patient is mitigated as fully as possible.
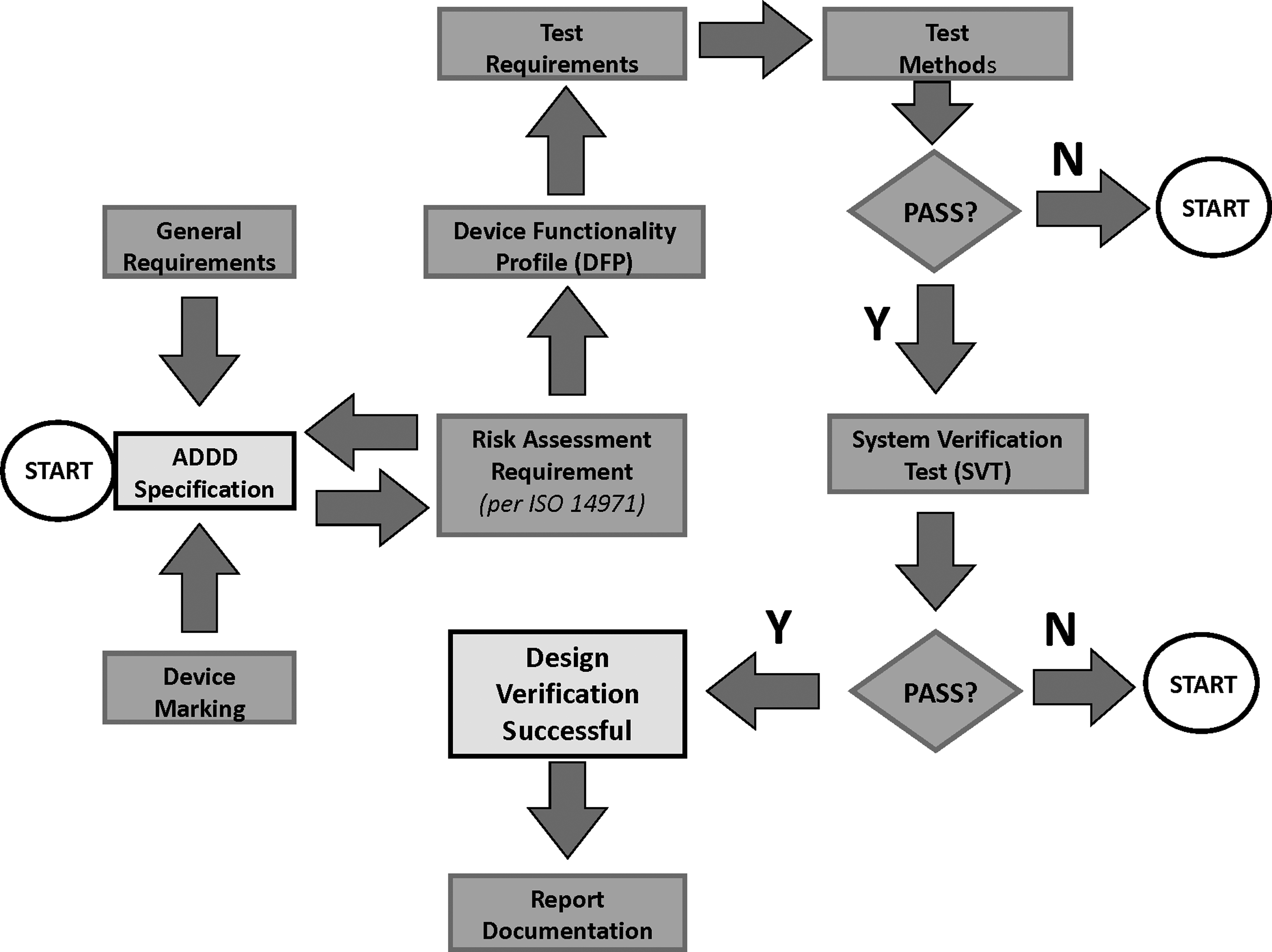
Process flow diagram showing the risk management approach to the design verification of an aerosol drug delivery device (ADDD)—adapted from ISO 20072:2009.
ISO 20072 specifically addresses the most basic elements regarding the safe and effective use of an ADDD, but it does not attempt to define the pharmaceutical or clinical performance of the finished drug–device combination. From a regulatory perspective, the ADDD design verification process is therefore the same, irrespective if the device is reviewed and approved as stand-alone unit, or if it is a part of a drug/medication system. It follows that after ISO 20072 has been applied to complete design verification of the device component, the normal regulatory process proceeds with the drug product–device combination in order to assure clinical safety and efficacy. However, if the approach advocated in ISO 20072 is followed and documented appropriately, it is anticipated that the following regulatory procedures should be more rapid, by simplifying the device approval part of the overall process. Within the EU, it should be noted that Annex 1 of the MDD(1) applies as far as safety and performance related to ADDD function.
ISO 20072 recognizes that the design verification process is a vital component of the overall validation process. The Device Functionality Profile (DFP) is the deliverable whose purpose is to define the parameters and tolerance intervals used to verify the ability of the ADDD to meet the manufacturer's design specifications during “in-use” conditions and also in a range of environmental/electromechanical “extreme exposure” conditions (Table 1). Examples of test procedures are given, but due to the vast number of device types encompassed within the scope of the standard, it is pointed out that it may be necessary to augment such methods with device-specific testing under certain circumstances where this approach is deemed appropriate. It is this nonrigid approach that will make ISO 20072 a potentially useful standard in the future development of OIP classes that fall within its scope. The standard also includes a system verification test (SVT) that should ideally be conducted at standard atmosphere and nominal airflow rates. This test comprises the measurement of emitted mass and, where appropriate, aerosol aerodynamic particle size distribution by the methodologies given in the pharmacopeial compendia. A suitable demonstration formulation chosen by the device manufacturer, which can be a placebo, representative medication or intended medication can be used for this test, which is intended to be conducted as the bridge between the device design process and likely performance of the finished product in use by a patient. The standard states that a subset of requirements can be applied in the early development of the ADDD, but all of the requirements must be satisfied as a part of the final design verification process. An open-ended and flexible approach to the prospect of new inhaler forms is adopted, by counseling that additional testing may be required when indicated by the risk assessment.
The underlying purpose of the 20072 standard is therefore that of a guide to methods for testing repeatability and reproducibility of ADDD functionality in a fully robust manner. Thus, guidance is therefore given on the number of ADDDs in the range from 15 to 60 to be evaluated in the development of the DVP for both “in-use” and “extreme condition” testing, thereby verifying the design intent to a high (95%) confidence level. Appropriate statistical methods are indicated to achieve this goal, incorporating a tolerance interval approach, rather than setting absolute limits for variability that would penalize the tester if more devices are evaluated. It should be noted, however, that the sampling plans do not replace assessment of the requirements associated with general manufacturing quality systems, which have to be addressed in the design process independently of the considerations associated with ISO 20072. This standard therefore enables the manufacturer to verify compliance with the design specifications thereby providing evidence to a Notifying Body (in the EU) or regulatory agency that user safety considerations have been addressed early during the development process. However, it has to be recognized that many nebulizer manufacturers may not have the internal capability to undertake the type of testing required to verify that the risk assessment called out in ISO 20072 has been satisfied. Under such circumstances, use of a third-party supplier of laboratory services with specific experience in testing OIPs would be a prudent decision to make.
ISO 27427:2010
The scope of ISO 27427 encompasses “general purpose” nebulizer systems intended for continuous or breath-actuated delivery of medication in aerosol form.(13) The term “general purpose” relates to nebulizers that are sold separately for use without being tied to a particular drug preparation for nebulization. The requirements for the safety, performance, and testing of these devices are specified in a prescriptive manner in ISO 27427. This approach is justified within the overall objective of ensuring suitability, safety, and component material compatibility with the dispensed liquid, together with an assessment of the biocompatibility of the materials used in manufacture of the nebulizing system. ISO 14971(16) is referred to in the context that nebulizing systems shall, when transported, installed, and operated in normal use and maintained in accordance with manufacturer instructions, cause no safety hazard that could reasonably be foreseen by a risk assessment in accordance with this standard.
However, beyond this general mention of the risk management approach, there is no attempt to mirror the development of a DFP and undertake a SVT at the design verification stage. The lack of a DFP with appropriate verification, as the core component early in the inhaler development process, makes the outcome of nebulizer development harder to predict, because it is up to each manufacturer to develop and implement their own interpretation of risk assessment and mitigation without the structured approach to the construction of the DFP afforded with ISO 20072. Furthermore, by placing much emphasis on performance verification of the finished product, manufacturers are more likely to test their new nebulizer toward the conclusion of development, when options to make improvements to the design are almost inevitably more restricted and usually more expensive to implement. Currently, the nebulizer manufacture intent on complying with the EN standard (or the ISO) standard need only follow a specific testing regime, and quote the test results in their sales and marketing literature. Often even this is beyond their internal capacity and under such circumstances, they should consider “contracting out” the tests to a third-party organization having the capability to both execute correctly the normative procedures provided by the standard for nebulizer performance testing and report the data in the way intended so as to provide a level playing field for clinicians to compare one nebulizer with another.
The test methods described in ISO 27427 are detailed to the extent that they include specifications for ancillary tubing, connectors to the nebulizing system, and the test equipment (filters, cascade impactor, test solution, etc.). However, instead of relying on the well-understood and validated methods for in vitro inhaler performance testing outlined in the pharmacopeias,(18,19) this standard describes a set of normative test methods for assessment of total output, output rate, and droplet sizing linked to operating attributes such as maximum/minimum fill volume, orientation, driving pressure, etc. (Table 2). There is an undoubted need to clarify the precise conditions under which the nebulizer should be operated and the metrics that are determined. However, by taking this approach toward providing detailed methodologies for executing the in vitro testing, the opportunity has been lost for harmonization with the new methods in clinically relevant ways that are about to be adopted, or are in the process of being adopted by the European and U.S. Pharmacopeias, respectively(20,21) (Fig. 1). Furthermore, validation of the test methods for a specific nebulizing system is not specifically mentioned in ISO 27427, increasing the potential for errors in the acquired data resulting from methodological bias. For example, the low flow impactor method that originated from EN13544:2001, involves anisokinetic sampling of the flow of nebulizer-produced aerosol (15 L/min) to the cascade impactor operating at a flow rate of 2 L/min. The process that is illustrated in which there is no on-axis removal of the sample stream to the impactor using a sharp-edged nozzle entry can result in significant size distribution bias, especially if droplets greater than 5 μm aerodynamic diameter are present.(22)
For gas-powered nebulizers, measurements to be made at minimum and maximum driving gas pressures and associated flow rates.
Fill volume is 2 mL or the volume recommended by the manufacturer.
A further concern associated with the prescriptive approach to the identification of factors associated with device safety, is the conceivable prospect that a manufacturer of a new type of nebulizing system in the future may not seek to assess and mitigate potential risks outside those that have been recognized in the standard. A relatively inflexible approach to performance verification compared with the way in which ISO 20072 has addressed this matter, may stifle development of novel technology where attributes are outside these norms. Such devices could therefore be difficult to verify as “safe” by a Notifying Body in the EU or its equivalent elsewhere. Had the determination of the operating “window” of the nebulizing system been adopted by ISO 27427 based on a systematic risk assessment based on all conceivable potential hazards, the former standard would have been more capable of adaptation as new technology is developed. Furthermore, it would have been a relatively simple task to harmonize to the corresponding approach taken in ISO 20072. As matters stand, some form of unspecified development risk assessment process is assumed to have taken place to resolve all design-related concerns, well before testing of the final product in accordance with ISO 27427, just prior to marketing.
This rigorous approach to testing, although laudable from the standpoint of being easy to follow in a step-by-step manner, will also make its adaption to nebulizing systems having nonstandard attributes difficult to perform, and the result may not be descriptive for the intended use. For example, unlike the 20072 standard where the manufacturer chooses a drug formulation that is representative in order to undertake the SVT, ISO 27427 specifies 1% w/v (10 mg/mL) salbutamol solution as the test formulation for all nebulizing systems. The question becomes: How suitable is this salbutamol solution for the liquid formulation (solution/suspension/emulsion/liposomal, etc.) for which a particular nebulizer may be specifically designed? Furthermore, 1% w/v salbutamol is not a standard concentration (i.e., 3 mL of a 2.5 mg/mL solution in the United States or 2.5 mL of a 1 or 2 mg/mL solution in the EU) currently marketed in ampoule form for use with nebulizers. Furthermore, the maximum concentration available in respirator solution contains only 5 mg/mL salbutamol, so the implication is that the tester will have to make up the solution from powdered salbutamol base or sulphate in physiologically normal saline. This is an unnecessary complication, given that accurately formulated solutions of this drug product are readily available. It is to be hoped that the current revision of this standard will enable these issues to be dealt with in a way that makes it easier for those making use of it to undertake testing specified.
Nebulizers are used with a wide variety of formulation types other than salbutamol solution, including suspensions of drug product in saline, emulsions, liposomal preparations, and high viscosity preparations for treating specific disease modalities. Physical characteristics, in particular density (of suspended particles), viscosity, and surface tension, all influence the aerosol particle size distribution.(23–26) Although ISO 27427 contains a “motherhood” clause: “[The manufacturer shall disclose in a] statement that using a solution, suspension or emulsion different from that recommended by the manufacturer, in particular for a suspension and/or high viscosity solution, may alter the particle size distribution curve,” no methodology is provided to assist the tester in establishing in precise terms how performance is affected. This situation is highly appropriate for a risk assessment-based approach, in which the manufacturer identifies potential formulations that might give rise to performance changes with nebulizer and conducts performance verification testing early in the development process.(24,27)
Summary and Conclusions
The development of the two ISO standards, 20072 and 27427, has been influenced differently due to the heritage and background of their sponsoring committees, as well as by differences in the backgrounds and balance of stakeholders involved with their inception. ISO 20072:2009 was developed without any previous influences that may have hindered the conception of the new and highly flexible approach that was adopted to derive the device DFP from a formalized risk assessment early in the development process at the design verification stage. On the other hand, ISO 27427:2010 evolved from a prior CEN nebulizer performance standard, with little further development. Currently, this standard is undergoing significant further revision in the light of numerous comments fed back from national standards bodies following the publication of the original version.(28) This process may result in important improvements to the more inflexible aspects associated with the current version. However, ideally, a more radical from-the-bottom-up revision is needed in which the design verification process is harmonized as fully as possible with that outlined by ISO 20072. There is no fundamental reason why nebulizing systems should be treated differently to other types of OIP when considering the process of going about design verification. It is noteworthy that ISO 20072 is unusual in that it does not have a “checkbox” approach for compliance. Instead, the expectation is that the manufacturer will develop an iterative approach from the outset (Fig. 2), systematically identifying risks that affect the critical aspects identified in the DFP, finding ways to mitigate them, and finally demonstrating that mitigation has been done as far as is possible, by testing that is appropriate for purpose and not merely a prescriptive method lacking flexibility. It is also recognized that given the diversity of devices that fall under this standard, it is neither possible nor desirable to try to cover and give prescriptive direction on what, how, when, and why to test in order to demonstrate compliance.
In overall terms, there is a clear tendency to incorporate risk management in the development of new ISO standards, a process that is in harmony with the Quality by Design principle that is increasingly being advocated by regulatory agencies, particularly in the United States. Given these circumstances, it is to be hoped that at a future revision of ISO 27427, the risk assessment approach, fully harmonized to that in ISO 20072, would be incorporated. Under such circumstances, the test methodologies outlined in 27427 could be retained as appendices, ideally harmonized with their equivalents in the United States and European Pharmacopeias, as they fulfill a role analogous with the in vitro testing chapters in the compendia.
An alternative, but more drastic solution, would be to unify the ideas from both ISO 20072 and 27427 into a single standard whose scope encompasses all OIP forms, utilizing appropriate compendial test methods as the means to determine performance characteristics. This approach could greatly simplify the process of compliance for manufacturers of nebulizing systems. Such harmonization might also result in cost reductions to the finished goods, by eliminating unnecessary and overprescriptive testing, an outcome that would be seen as positive by stakeholders involved with the payment for these devices.
Footnotes
Author Disclosure Statement
Neither author has any commercial conflict of interest to declare in connection with this article. Jolyon Mitchell served on the committees that produced both ISO 20072:2009 and ISO 27427:2010.