
Abstract
Abstract
Regulatory guidance in Canada and Europe recommends that the manufacturer of an inhaled drug product delivered by pressurized metered-dose inhaler (pMDI) identify a spacer (S) or valved holding chamber (VHC) to be used with their designated product. It therefore becomes necessary to include the S/VHC in the process of establishing bioequivalence (BE) to the reference pMDI product for both new-entry generic and subsequent market entry products (SMEPs). S/VHCs substantially modify the aerodynamic particle size distribution (APSD) of the inhaled medication, and potentially the spatial distribution of the mass of active pharmaceutical ingredient(s) [API(s)] depositing in the respiratory tract. The processes whereby S/VHCs can influence BE outcomes are examined, and the inadequacy of compendial in vitro methods to provide pertinent information to assess BE for the pMDI+VHC combination is highlighted. A three-part strategy is proposed whereby in vitro testing for BE can simulate more clinically-relevant conditions than in the current compendial procedures:
1. The inclusion of a short delay between inhaler actuation and sampling onset is appropriate when determining APSD at flow rate(s) suitable for the intended patient population; 2. Assessment of total emitted mass ex S/VHC by simulating tidal breathing pattern(s) appropriate for intended use; 3. Incorporation of appropriate face model(s), representative of the intended patient age range(s), into test procedures for S/VHCs with facemask, enabling clinically-appropriate dead space and fit-to-face to be simulated.
Although the compendial authorities have been slow to recognize the need for such in vitro testing, a Canadian standard provides direction for implementing most proposals, which should result in better performance predictions and more appropriate clinical outcomes, highlighting similarities and differences between reference and test products.
I. Introduction: Bioequivalence, Relative Potency, and the Delivery of Medication to the Lungs by Pressurized Metered-Dose Inhaler
Bioequivalence (BE) in the context of comparing generic and subsequent market entry products (SMEPs) with an innovator OIP can be defined by reference to a hypothetical example of a plot of dose of active pharmaceutical product (API) received by the patient versus an appropriate measure of clinical response for two different OIPs in the same therapeutic class (Fig. 1). In the absence of any information concerning the position of the dose–response curves for these products, the “test” product (OIP1) would be deemed bioequivalent to the “reference” product (OIP2) when the clinical outcome is the same for a given dose, in this example, Dose C. Additionally, in the example shown, the dose–response curve for OIP2 is displaced to the right of that for OIP1, reflecting a difference in relative potency (RP) between the two products: the same clinical response is elicited at Dose A for OIP1 and Dose B for OIP2, and therefore, by definition, the RP for the two products is the dose ratio B/A. It is important also to note that likely the clinical response for both products will reach a plateau above which further increases in dose (that is, greater than Dose C) will fail to elicit further improvement in response. In summary, at dose level “C” in Figure 1, both products would be deemed bioequivalent, despite having different RPs. Regardless of whether or not an S/VHC is being assessed with test and reference pMDI products, it is necessary to undertake clinical trials assessing RP at dosing levels for both products well below the plateau region to provide adequate sensitivity to any change in dose delivered to the patient.(3) The use of test doses at the “plateau” of the dose–response curves is likely to demonstrate equivalence between test and reference products, but mask real differences in performance.
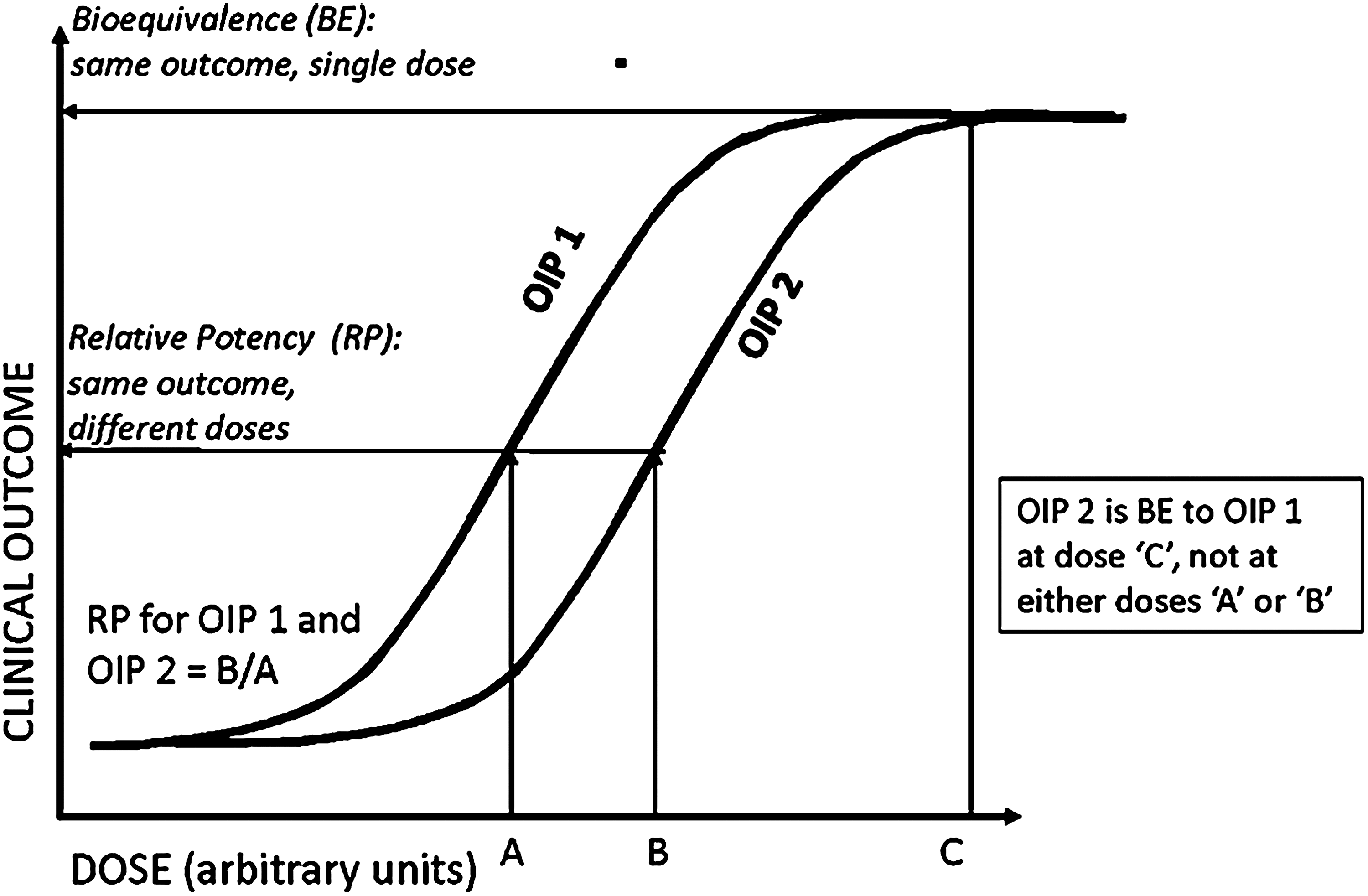
Definitions of relative potency and bioequivalence for two hypothetical OIP products in the same therapeutic class (adapted from Parameswaran et al.(140)).
The addition of the S/VHC also broadens the procedure when establishing RP and BE, by providing two options in clinical use: the pMDI alone and the pMDI with S/VHC. At its most extreme, the task now becomes a twofold process ultimately involving as many as four comparisons (Fig. 2):
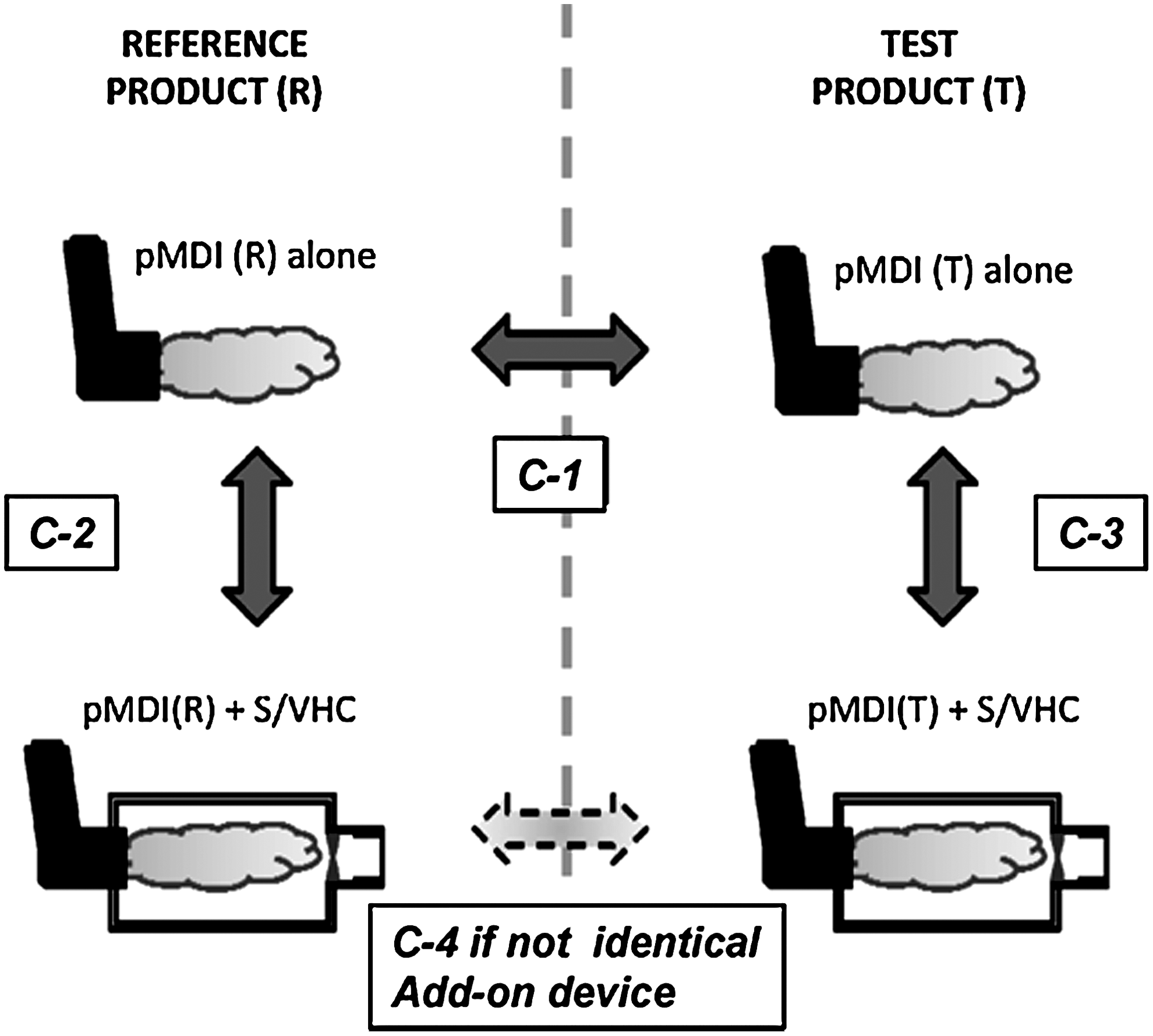
Three possibilities for BE testing for pMDI products with S/VHC add-on devices.
Test C-1 for the test and reference pMDIs without an add-on device;
Tests C-2 and C-3 for reference and test pMDIs, respectively, each with the nominated S/VHC.
If tests C-1, C-2, and C-3 all demonstrate equivalence between test and reference conditions, then it can be assumed that the combination pMDI(R)+S/VHC is also equivalent to pMDI(T)+S/VHC, as long as the S/VHC is the same device. In the unlikely event that this is not the case, then it will be necessary to make the additional comparison, identified as C-4.
Looking at the information in Figure 2 in more detail, C-1 is the comparison that is already undertaken for test and reference inhaler products without S/VHC, and provides a reference point for those familiar with pMDI-to-pMDI comparisons in the context of the product approval process for a generic pMDI. Pathway C-2 represents the comparison between the reference pMDI and its nominated S/VHC on the Summary of Product Characteristics (SmPC) in Europe. The comparison when an S/VHC is involved is more complex than the pMDI-to-pMDI comparison, because the add-on device modifies the emitted aerosol by largely eliminating the ballistic fraction that comprises most of the coarse particle mass fraction of the dose that would otherwise deposit in the oropharynx. In addition, this portion of the emitted mass may have both topical (i.e., oral candidiasis in the case of ICS(4)) and systemic effects, which are dependent on the metabolic pathway of the API(s) associated with the formulation, through swallowing and subsequent absorption via the gastrointestinal tract.(5) It is therefore envisaged that the comparison for equivalence will be based on matching primarily the therapeutically-beneficial portion of the dose (i.e., the fine particle fraction including or excluding extra-fine fraction, if, in the latter instance, it is believed that the extrafines are inhaled but do not deposit in the lungs(6)) as a measure of clinical efficacy. It is further foreseen that the comparison C-3 would apply in the case of a second-entry pMDI where the innovator product already has an S/VHC specified on its SmPC. Comparison C-3 would again be based on the fine/extra-fine portions of the dose, with the objective of matching with that emitted by the test pMDI, and ultimately to the reference pMDI through comparison C-1. However, the S/VHC for the second entry pMDI need not necessarily have to be the same design as that specified in the SmPC of the innovator product. If this is the case, then potentially a fourth comparison (C-4) could be made between the test and reference pMDIs, each with its respective but different S/VHC.
The same regulatory agencies in Europe and Canada have accepted that in vitro data may be used in support of arguments for BE.(1,2) However, it is recognized that establishing valid statistical criteria for demonstrating equivalence is challenging,(7–9) particularly in the context of comparing complete aerodynamic particle-size distribution (APSD) data from cascade impactor measurements that are considered to provide meaningful in vitro measurements predictive of particle deposition in the human respiratory tract.(10) Comprehensive research on this topic was undertaken in the mid 2000s by a working group of the US-based Product Quality Research Institute (PQRI) on methods based on chi-square analysis of mass distribution profiles from a database of APSDs derived from most classes of OIPs,(7) supplemented by a population bioequivalence (PBE) test for impactor-sized mass (ISM).(8) They concluded that in some situations the chi-square ratio test alone was sufficient to determine equivalence of APSD profiles. However, in many instances, the combined application of these tests appeared to increase the discriminating ability of the statistical procedure compared with the chi-square ratio test alone. Yet, in other situations, neither of the tests alone nor their combination was adequate.(9) At the present time, there therefore appears to be no simple statistical test that has universal capability to define in vitro equivalence for the inhaler alone, not including the complicating factor of the presence of an add-on device that substantially modifies the pMDI-emitted APSD.(3) Furthermore, the relationship between metrics obtainable from laboratory testing, such as coarse and fine particle dose (mass) based on either nominal (label claim) dose or the actual emitted dose from the OIP mouthpiece, and clinical response, is complex (Fig. 3). This association is influenced both in vivo by partitioning of the inhaled API between the respiratory tract and the gastrointestinal tract and in vitro by the efficiency of the OIP as an aerosol generator. Both types of partitioning, as well as the proportion of the inhaled dose that reaches the lungs compared with that depositing in the oropharynx (or nasopharynx for obligate nose-breathing patients), are substantially affected by the presence of an S/VHC.(3)
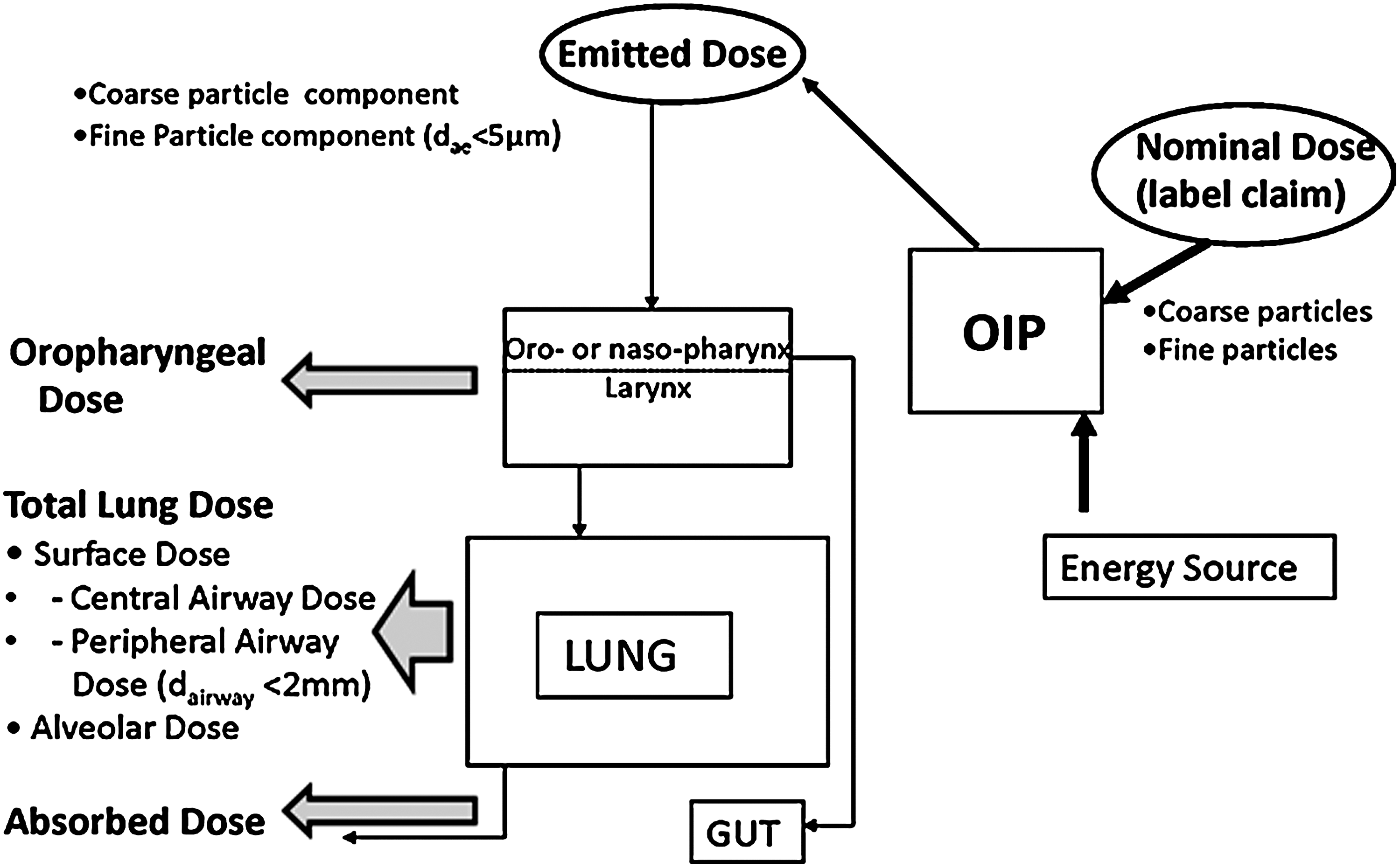
Physical and biological processes associated with delivery of inhaled medication to the lungs (adapted from Dolovich(141)).
The issues surrounding pMDI-to-pMDI comparisons have been reviewed in a workshop organized by PQRI in 2009.(11) The specific role of pharmacokinetics in establishing BE for OIPs was more recently assessed in a further PQRI-sponsored workshop that took place in conjunction with the Respiratory Drug Delivery conference in 2010.(12) As the focus of both workshops was largely on the OIPs themselves (comparison C-1 above), the outcomes will not be discussed in this article. Neither will statistical considerations associated with these comparisons, although it is recognized that ultimately progress will need to be made to establish robust statistical criteria, even if they have to be tailored for specific inhaler classes or APSDs. It is worthwhile noting that the 2009 PQRI consensus report made the important observation that as pMDIs can be used in combination with ancillary devices such as spacers (and VHCs), consideration needs to be given to the impact of the (add-on) device design on the performance of the pMDI-ancillary device combination.(11) Thus, there is a growing understanding among regulators that, in considerations of BE, the pMDI+add-on device combination should be considered as an inhaled medication system in its own right for the delivery of the API(s) associated with the OIP.
The US position with respect to the process of demonstrating BE for OIPs is set out in the Code of Federal Regulations under regulation 21 CFR 320.24(b).(13) This document allows the following testing options to be considered by the sponsor of a new drug application (NDA):
1. in vivo studies in humans comparing drug-metabolite concentrations in an accessible biological fluid;
2. in vivo testing in humans of an acute pharmacological effect;
3. controlled clinical trials in humans to establish safety and efficacy;
4. in vitro methods;
5. any other approach deemed adequate by Federal Drug Administration (FDA).
There is currently no specific mention of requirements related to S/VHCs in this regulation. However, in a draft FDA Chemistry, Materials and Controls industry guidance for OIPs issued in 1998 by the Center for Drug Evaluation and Research (CDER), there is recognition that clinical efficacy assessment of pMDI-delivered aerosols requires consideration of the presence of an S/VHC.(14) This position contrasts with the aforementioned European and Canadian regulatory guidance documents that make specific mention of the need to consider in submissions the clinical performance of the nominated add-on device.(1,2)
Although often simply referred to as “spacers,” a number of current European and Canadian clinical guidelines recommend the use of VHCs rather than open-tube spacing devices for the treatment of asthma and chronic obstructive pulmonary disease (COPD), especially for small children and the elderly, as open-tube spacers are unable to retain the aerosolized medication.(15–20) This article therefore focuses mainly on the VHC as the preferred pMDI add-on device, in relation to the testing procedures that might be adopted to compare performance between delivery systems.
Metrics That Are Useful in Evaluating VHCs in the Laboratory
Before addressing how the addition of a VHC might influence in vitro measures of performance to support clinical assessments of BE, it is useful to review the choice of measurement apparatus to obtain meaningful particle size-related information, together with the derivation of the metrics that are of most use to the process.
It is important to appreciate that the primary purpose of the pharmacopeial laboratory evaluation methods for dose-content uniformity and APSD is one of product quality control in the context of batch release.(21) Under these circumstances, the ability of the test to discriminate between “good” and “poor” product is paramount. It therefore follows that these tests have been designed to be as simple as possible to carry out, in the interest of minimizing contributory causes to method variability.(22) In consequence, refinements such as the use of breathing simulation and the mimicking of the uncoordinated user, which might be expected to provide data that are more closely representative of clinical use,(23) are absent. It is our belief that the incorporation of these more clinically appropriate methods for laboratory testing of these devices, as outlined later in this article, will do much to provide measures of in vitro performance that are more easily related to particle deposition profiles in the respiratory tract and therefore to clinical outcomes.
Although laborious and complex in terms of the sources of variability,(24) the multistage cascade impactor remains the mainstay for OIP APSD assessment.(25) This is because these apparatuses directly link the determination of mass of API in the OIP aerosol with the aerodynamic size scale that is pertinent in terms of predicting deposition of aerosol particles in the respiratory tract.(26) For this reason, the cascade impactor has been accepted by both the compendial and regulatory authorities as the apparatus of choice for sizing these aerosols in the context of VHC assessments.(27–29) Alternative techniques, such as time-of-flight aerodynamic particle size analysis, can size OIP aerosols in terms of aerodynamic diameter more rapidly than the CI method.(30) However, these instruments do not provide traceability to the mass of API(s) in the formulation through the use of well-defined and widely accepted analytical assay principles (i.e., high-performance liquid chromatography combined with a quantitative spectroscopic detection method, usually UV/visible light absorption or fluorescence, which is API-specific).(30) API traceability is regarded as being of critical importance for in vitro assessments of OIP performance characteristics by the major regulatory agencies in Europe and North America.(27–29) Similar considerations apply to laser diffractrometry (LD), but, in addition, this technique does not measure particle aerodynamic size. Instead, LD determines a measure of size that is related to the parameters linked to the physical dimensions of the particles, loosely associated with volume equivalent diameter.(31) Other non-cascade impactor techniques are also subject to one or the other or both of these restrictions.(32)
Figure 4 illustrates schematically the overall effect of a VHC of non-specific design on the emitted aerosol, and Table 1 lists the API mass-based metrics that can be determined from a cascade impactor-based method. Later in this article, it will be shown that testing of these devices by breathing simulator is also necessary to support verification of the function of both the inhalation and exhalation valves (the latter being used with the facemask patient interface to permit tidal breathing without the need to remove the facemask). The few additional measures specific to this type of testing are useful to augment cascade impactor-based measurements, specifically in relation to the impact of poor patient coordination on VHC performance.
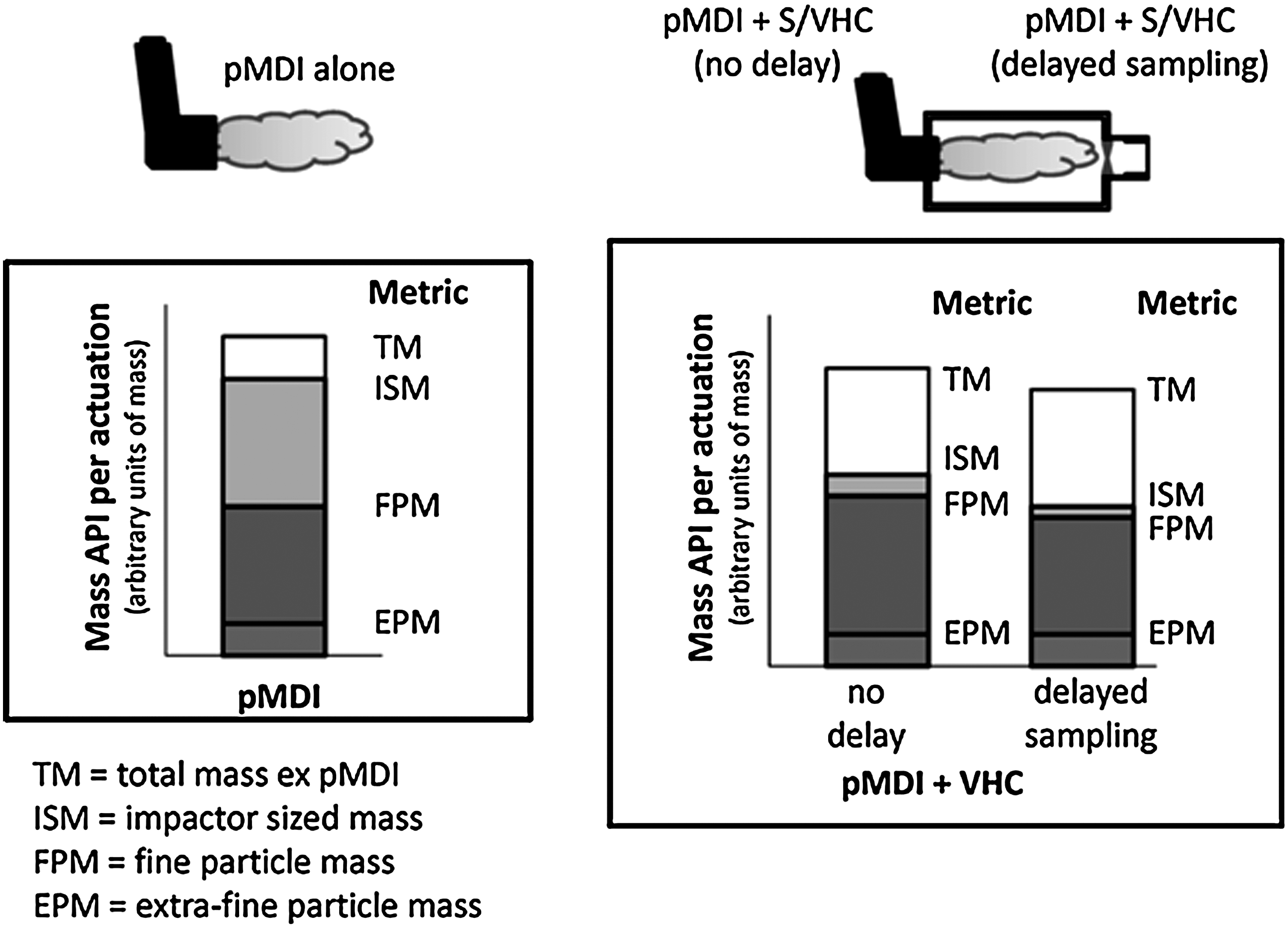
Metrics associated with laboratory performance testing of VHCs by the cascade impaction method.
Based on mass/actuation—more than 1 actuation will probably be needed for adequate assay of the API.
The delay time is stated in seconds.
Measurements of Emitted Mass of API from the Inhaler
From closer inspection of Figure 4, it can be seen that the total mass of API emitted from the inhaler mouthpiece (TMMDI) is modified by the mass lost to the interior surfaces of the VHC, so that the total mass of API emitted from the VHC without a delay interval (TMMDI+VHC-0s) will always be less than TMMDI. It should be noted that there can never be any delay imposed between inhaler actuation and the onset of sampling when the pMDI is evaluated alone, or else the aerosol would be lost to the sampling system. When a VHC is added, there is the option to sample the aerosol with or without a short delay following inhaler actuation, so the subscript extension “_s” used in Table 2 and elsewhere in this article represents the delay interval in seconds between pMDI actuation and onset of sampling.
TMMDI+VHC-0s is the measure of API mass in the no-delay condition that is emitted at the mouthpiece or available to be inhaled at the facemask of the VHC, depending on which patient interface is present. Delayed sampling from the VHC will further reduce TM MDI+VHC as the result of time-dependent aerosol depletion processes taking place within the add-on device.
ISM represents the mass of API that penetrates as far as the first impaction stage with a defined upper boundary size limit. In the Andersen 8-stage cascade impactor (ACI) that is widely used for OIP assessments,(25) this is stage 1 in the configuration that is used for pMDI testing, whose upper boundary size is 9.0 μm aerodynamic diameter at 28.3 L/min.(25) The upper boundary size is undefined for stage 0 in this configuration, so that the usually small mass of API collecting on stage 0 is normally assigned to the coarse particle mass (CPM; not shown separately in Fig. 2). ISM together with the mass on the stage 1 is sometimes referred to as impactor mass (IM). CPM therefore mainly comprises the mass of API recovered from the induction port. In the Next Generation Pharmaceutical Impactor (NGI), the first impaction stage (stage 1) has its upper boundary size defined by archival calibration with standard-sized particles,(33) so that CPM is just the mass retained by the induction port and ISM, defined as the cumulative mass collected from this stage to the micro-orifice collector or backup filter, is identical with IM.
FPM and EPM represent the mass of API associated with the two subfractions of the emitted aerosol with the potential for therapeutic action. The cumulative mass-weighted APSD, as shown by the generic example in Figure 5, enables the corresponding mass fractions, FPF and EPF, to be obtained as percentages of either ISM or TM. Normalizing these mass fractions with respect to ISM or the mass entering the impactor (IM) can be helpful if the shape of the APSD is of interest from one pMDI+VHC comparison to another. However, TM is more useful than either ISM or IM as the reference metric in calculations of these mass fractions, because it allows for the impact of the VHC on CPM to be made evident in comparisons with the pMDI alone (C-2 and C-3 in Fig. 2).

Metrics associated with laboratory testing of pMDI-VHCs by a cascade impaction method. The elimination of the ballistic fraction ejected by the inhaler results in an increase in FPF<4.7μm aerodynamic diameter.
In European submissions for new pMDI-based products based on monograph 2.9.18 of the European Pharmacopeia, the FPF metric is related to particles finer than 5.0 μm aerodynamic diameter (FPF<5μm) as the surrogate for the portion of the dose that reaches the lower respiratory tract.(34) For Canadian and US submissions that are based on the methodology provided in chapter 601 of the US Pharmacopeia (USP), the upper size limit for FPF is currently undefined.(35) However, it is common practice to choose 4.7 μm aerodynamic diameter, as this upper limit corresponds to the cut-point size of stage 2 of the ACI (Apparatus 1 of the USP), when sampling at 28.3 L/min.(25) FPF may also be defined with both upper and lower boundary sizes, typically by grouping the mass of API that collects on the appropriate intermediate stages of the impactor. In the example shown in Figure 5, FPF defined in this way is calculated from the sum of masses collecting from stages 3 to 5 in the ACI. Whichever approach is chosen to define FPF, the hypothesis is made that FPM, calculated as the product of FPF and TM, is related to lung deposition and, therefore, ultimately to appropriate measures of clinical efficacy, depending on the therapeutic class of the API.(21)
How VHCs Can Affect Bioequivalence
Spacers and VHCs are prescribed primarily for infants and small children(36,37) and also for older patients that have poor compliance associated with imperfect coordination of pMDI actuation and inhalation, including conditions that impair their ability to use a mouthpiece.(38) These devices interface directly with the actuator/mouthpiece of the pMDI, primarily providing a conduit with specified additional volume for the aerosol plume to develop.(36) Spacers are open tubes, lacking a valve that would prevent aerosol escaping before the user is able to inhale.(39) As such, their primary function is to extend the distance between the inhaler and the patient's mouth. Inadvertent exhalation into the open tube spacer will therefore result in loss of medication through the air inlet(s) at the spacer distal end.(39) VHCs, on the other hand, contain as a minimum, a one-way valve at the patient interface that allows the aerosol plume to be retained until it can be inhaled.(40) In consequence, VHCs rather than spacers are to be preferred, particularly for younger pediatric patients,(40) given the need for similar coordination ability as if using the pMDI alone. Once the aerosol plume arrives in the VHC, other than particle losses that deplete the ballistic fraction due to inertial impaction on the interior surfaces, the remaining aerosol is subjected to several forces that will tend to deplete its concentration with elapsed time (Fig. 6).(41) These are most importantly gravitational sedimentation and electrostatic charge (if not mitigated by the use of charge dissipative/conducting materials or by pretreatment using a washing procedure involving detergent). Consideration of such time-dependent removal processes will therefore be important when delayed inhalation is simulated as part of the laboratory test program in support of BE for these devices.
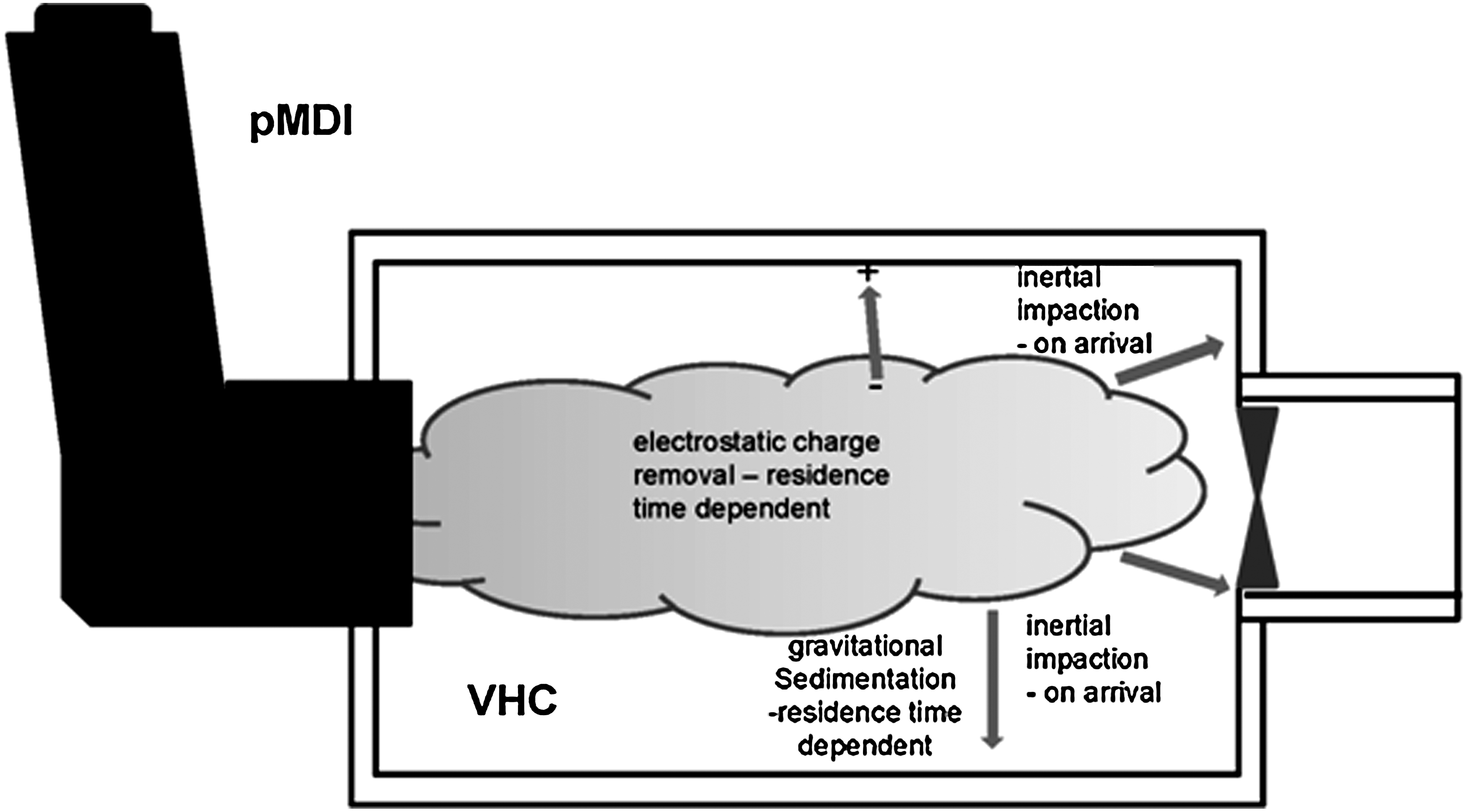
Aerosol depletion processes in a VHC. Inertial impaction of the ballistic fraction to the walls on arrival and continuous removal by gravitational sedimentation subsequently dominate aerosol mechanics if electrostatic charge-related effects are eliminated.
The following physical processes are important in governing the behavior of a pMDI-generated aerosol that is confined within a VHC as part of the medication delivery system.
Removal of the ballistic fraction from the pMDI
A principal function of the VHC is the removal of the pMDI-generated ballistic component comprising the large droplets ejected from the inhaler by flash-evaporation of the propellant.(42) This process takes place before this fraction of the total mass of API ex metering valve can reach the airway of the patient.(43) Removal occurs primarily as the result of inertial impaction and turbulent deposition of the spray droplets on the distal valve and interior walls of the VHC combined with further evaporation of liquid propellant and low volatile cosolvent, if present.(44) The resulting effect of these processes on the measured cumulative mass-weighted APSD determined by the cascade impaction method is exemplified by data from a commercially available hydrofluoroalkane (HFA)-suspension product in which salbutamol was the API (Fig. 5). Note that the APSD of the nonballistic fraction sized by multistage cascade impactor is comparable with or without the VHC present, when the data from the pMDI alone are scaled in terms of ISM. The loss of the ballistic portion in the VHC shifts the APSD of the aerosol scaled in terms of TM in the direction of finer sizes compared with that for the pMDI alone, so that FPFMDI+VHC is substantially increased compared with FPFMDI. Thus, use of the VHC results in a slower-moving aerosol that is available for inhalation and comprised of smaller particles that have a greater probability of penetrating to the lower respiratory tract. The spatial distribution of deposited particles in the lungs, containing the API or APIs (in the case of a combination product), is therefore substantially altered by the presence of the add-on device.(45) This phenomenon has also been demonstrated on many occasions by gamma-scintigraphic studies with patients inhaling medication via various pMDI+VHC combinations, where the goal has been to assess lung deposition as a surrogate for clinical efficacy.(46) Lung deposition from these add-on devices depends not only on their physical characteristics (primarily internal volume and shape(43)) but also on age-specific inhalation profiles and airway morphology, including disease-associated changes.(45) Given this dependency with respect to such patient-specific characteristics, the need for in vitro testing for the assessment of BE, under conditions that more closely simulate actual use, becomes self-evident. This link is justified on the basis that clinical efficacy, and therefore BE metrics, are ultimately related either to the spatial deposition characteristics of inhaled APIs to receptors located in the lower respiratory tract for topical delivery, and/or to the small portion of the inhaled mass of API delivered to the alveolar spaces representing systemic delivery of medication.(45,46)
Electrostatic charge
pMDI-generated aerosols acquire intrinsic electrostatic charge by the process of triboelectrification that takes place during aerosol plume formation.(47,48) VHCs made from nonconducting polymers also acquire surface electrostatic charge during manufacture, packaging, and storage.(41,49,50) The presence of electrostatic charge from either cause results in reduced and more variable medication delivery,(51) making it difficult to undertake reliable comparisons in connection with BE. It is therefore important, in the in vitro assessment of VHC performance, that precautions are taken to mitigate charge if the add-on device is manufactured from nonconducting materials. This process can be readily accomplished by prewashing with mild detergent followed by drip-drying in air.(52) Alternatively, the problem can be avoided altogether by specifying the VHC as one manufactured from either charge-dissipative(53) or electrically-conducting(41) materials. The use of these materials in VHC construction also avoids the risk that the user of a nonconducting device may fail to carry out pretreatment following the manufacturer's instructions, possibly resulting in variable dosing.(54)
Gravitational sedimentation
The settling of individual aerosol particles in the plume that is retained by the VHC is a time-dependent process governed by the gravitational force that is continuously present.(41) If electrostatic interactions between particles and the interior of the VHC are eliminated, once any turbulence associated with plume entry dissipates, sedimentation following Stokes's law becomes the dominant process that preferentially removes the largest airborne particles that are present.(55) The length of the delay period before inhalation takes place influences the proportion of the dose from the pMDI that is eventually delivered to the mouth, for a given internal geometry. As a rule, VHCs with a greater internal diameter will result in an increased proportion of inhalable particles of a given size that remain airborne after a given delay interval.(41) Settling velocities in air at atmospheric pressure for fine particles less than 5 μm aerodynamic diameter are less than 7.6×10–2 cm s–1. Smaller and near-to-ideal capacity VHCs of ca. 150 mL(43) have radii that are typically about 2 cm (assuming the add-on to be cylindrical and oriented horizontally (i.e., the most favorable direction for sedimentation-induced particle loss). A 5-μm unit density (c.g.s. system) particle located along the central axis of the VHC could ideally take about 26 sec to deposit by sedimentation alone to the VHC wall. However, rather than calculate individual particle size–related settling times to arrive at a measure of aerosol retention, it is easier to consider the half-life (t½), or the time to deplete 50% of the retained aerosol mass. As an example of what might be expected with actual VHCs, Bisgaard reported t1/2 values that were in excess of 30 sec for pMDI-delivered chlorofluorocarbon (CFC)-formulated budesonide aerosol delivered via a pear-shaped electrical conducting, horizontally-held 220-mL VHC.(56) This t1/2 compared with 9 sec for a similarly oriented 750-mL elongated ellipsoidal shaped VHC, in which surface electrostatic charge had been eliminated as a confounding factor.(56) The FPF<4.7 μm aerodynamic diameter was reported as being between 69% and 70% in another related study, sampling the aerosol from these devices with no delay between inhaler actuation and onset of sampling.(57) It is therefore likely that even longer t1/2 values would be obtained with the finer aerosols that are more typical of current HFA-formulated products.(58,59) As the particle-settling characteristics are governed by the design (capacity and internal geometry) of the particular VHC as well as the APSD of the retained aerosol, it follows that similarity in both shape and capacity between test and reference VHCs will be important in comparisons for BE (Fig. 2).
Particle-size shifts caused by enhanced evaporation of cosolvent
A both serendipitous and important development adopted by the pharmaceutical industry, in response to the transition from CFC to HFA propellants, was the incorporation of ethanol as a cosolvent for the API, specifically for dispersing many of the glucocorticosteroids with low solubility in the available HFA propellants.(58) The first solution-formulated product, Qvar, developed by 3M Pharmaceuticals (St. Paul, MN), was intended to deliver a large proportion of the dose of beclometasone dipropionate (BDP) topically to the distal airways of the lungs as extra-fine particles.(59) In vitro studies by ACI characterized the mass median aerodynamic diameter (MMAD) of the emitted aerosol to be close to 1.1 μm, almost threefold smaller than the emitted aerosol from the original CFC-based suspension product.(59) This reduction in MMAD was associated with a shift in the entire APSD sized by the impactor to finer sizes. However, Stein(60) observed that the ethanol cosolvent associated with the largest droplets emitted from the inhaler can take several seconds at room ambient conditions to evaporate completely after the aerosol plume is generated. This is a relatively lengthy evaporation process, compared to the evaporation time for the sub-10-μm fraction in transit from the inhaler mouthpiece to the induction port inlet as far as the impactor entry, which can be <50 ms.(61) The effect of this process on aerosol measurement of the aerosol emitted from Qvar pMDIs (formulated with 8% wt/vol ethanol) without a VHC was recently demonstrated in the laboratory using liquid ethanol-sensitive filter paper.(62) Liquid droplets were found to be still present after aerosol had been transported from the inhaler mouthpiece through the ca. 200-mL internal volume of the USP/Ph.Eur. induction port, reaching as far as the uppermost impaction stage of an abbreviated ACI. Given the relatively slow evaporation of ethanol in the largest droplets emitted by this OIP, the insertion of a VHC will allow extra time for the evaporation process to proceed toward completion before measurement. In the context of in vitro testing for BE considerations, it is useful to consider what might happen to the resulting aerosol while retained by the VHC before being sampled, especially if delayed inhalation is being simulated as recommended by the EMA Guidance.(1) This question was addressed by a laboratory study by Mitchell et al., in which small-volume VHCs (149 mL) were compared with two slightly different HFA solution formulations (Qvar, TEVA Pharmaceutical Industries Ltd., 100 μg of BDP/actuation; and Alvesco, Nycomed Canada Inc., 100 μg of ciclesonide).(63) In the benchmark measurements with no delay between inhaler actuation and the onset of sampling, simulating use by a perfectly coordinated patient (Table 3), fine particle mass (FPM<4.7μm) per actuation less than 4.7 μm aerodynamic diameter [63.1±6.0 μg (Qvar); 68.2±1.8 μg (Alvesco)] via an antistatic VHC increased with both drug products compared with testing the pMDI alone [41.9±2.2 μg (Qvar); 42.4±3.8 μg]. When a more realistic 2-sec delay was introduced to mimic the more likely case of use by a poorly coordinated patient,(64) the corresponding values of FPM<4.7μm from the different VHCs were 44.4±4.5 μg and 51.5±3.9 μg for Qvar and Alvesco, respectively. These data were much closer to FPM determined from either pMDI when used without a VHC. The overall APSD of the aerosol entering the impactor was observed to be only slightly narrowed by the presence of the VHC when delayed sampling took place, as shown by the data for Qvar that are illustrated in Figure 7. In passing, it is worth noting that Dolovich earlier observed with aerosols from several different CFC-formulated pMDIs that the presence of a VHC could increase the ratio of fine particle mass compared with that emitted by the pMDI alone by more than 20%.(64) The cause of this behavior is almost certainly associated with mass transfer of particles, which would otherwise be collected in the coarse fraction, to the fine particle fraction as the result of remaining propellant evaporation in the additional space afforded by the VHC. However, Harrison has suggested that the breakup of larger BDP particles could also contribute to the higher than expected FPM with no delay following inhaler actuation.(65)
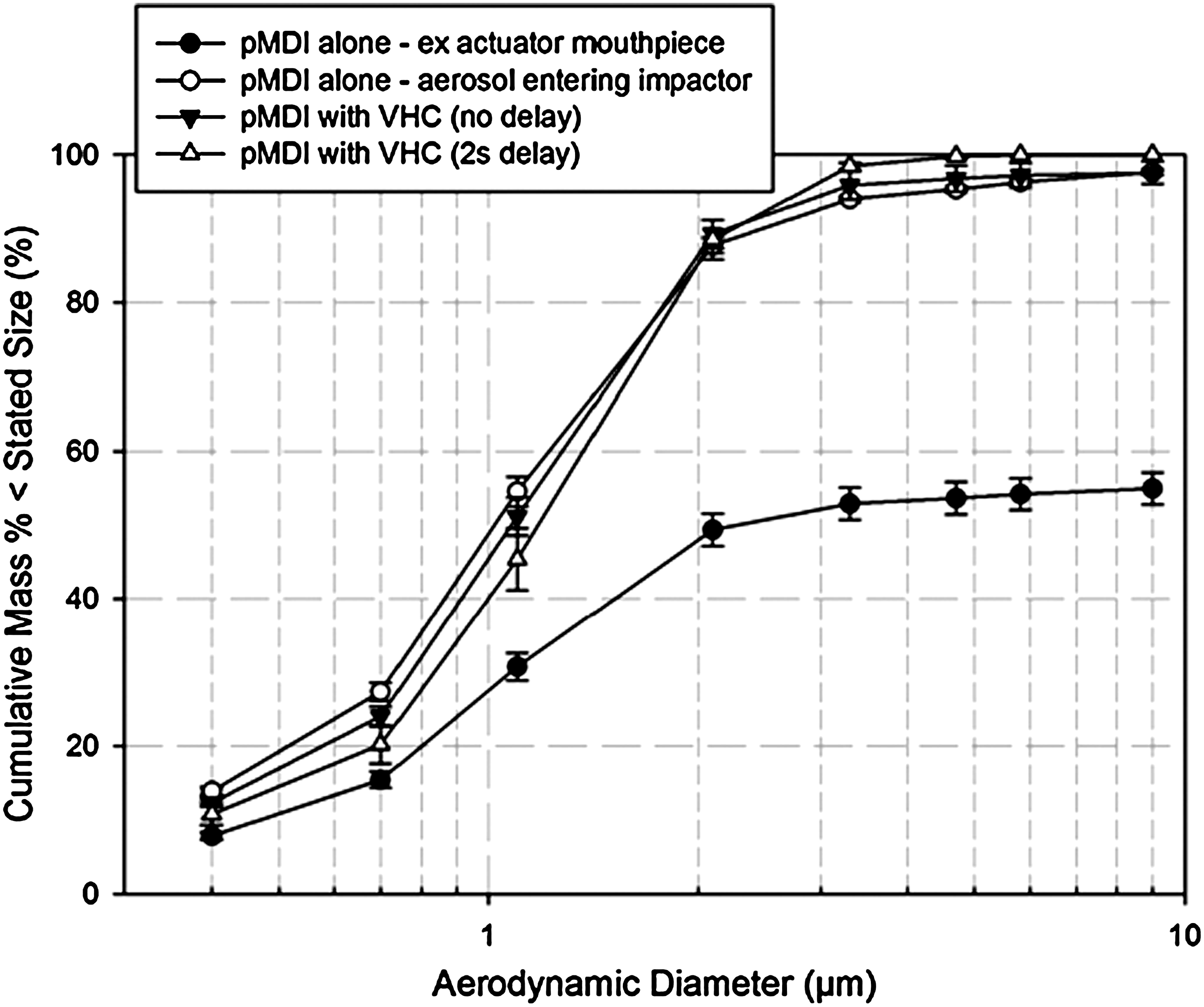
ACI-measured APSDs for Qvar, a formulation containing 8% wt/vol ethanol as low-volatile cosolvent.
Data are from Ref. 53.
FP, fluticasone propionate; SX, salmeterol xinafoate; BUD, budesonide; FF, formoterol fumarate.
The study of Mitchell et al.(63) was designed to demonstrate comparability for in vitro performance with a wide range of different drug products between the three versions of the same 149-mL cylindrical VHC that were available worldwide in 2009. The data in Table 3 (mean±SD) confirm the substantial similarity in FPM<4.7μm between VHCs manufactured from charge-dissipative materials and those from nonconducting polymers following pretreatment to eliminate electrostatic charge with a 2-sec delay between inhaler actuation and the onset of sampling and FPM<4.7μm from the pMDI alone. However, this investigation did not go further to examine the effect of chamber volume on FPM. It is known that larger VHCs may deliver more medication in the form of fine particles from solution formulations than would be the case from the pMDI alone. The underlying cause again appears to be associated with more space for the aerosol plume from the pMDI to occupy and therefore longer time for the ethanol to evaporate before being inhaled from the VHC.(66) In the present context of investigating the properties of a VHC that effect BE, it would therefore be a useful exercise to repeat the work of Corr et al.,(43) which was undertaken in the early 1980s, using CFC-based pMDIs, with selected currently available HFA-formulated solution and suspension formulations.
Although Mitchell et al. reported that FPM<4.7μm ex VHC was increased in the no-delay condition with the solution formulations (Alvesco and Qvar) compared with the equivalent values obtained for the pMDI alone,(63) it is interesting that the APSDs of these impactor-sized aerosols hardly changed (see Fig. 7 for Qvar). It is therefore plausible that cosolvent evaporation may be of little consequence as far as modifying the APSD of these aerosols available to be inhaled from these particular-sized VHCs either with no delay or if the delay is ≤2 sec. Nevertheless, it is germane to question whether the slightly increased values of FPM ex VHC observed in this laboratory-based study with no delay compared with that from the pMDI alone might result in measurably greater clinical efficacy, given that patients rarely have perfect coordination between pMDI actuation and the onset of inspiration. The recent gamma-scintigraphic study with 99m-technetium-radiolabeled BDP (Qvar) by Leach and Colice(67) with similar-sized VHCs appears to provide at least partial insight. This group evaluated particle deposition in 12 adult healthy and inhaler-trained subjects (three subjects per study arm) who inhaled a 50 μg/actuation dose ex metering valve of the pMDI, followed by breath-holding of varying duration. In another arm of their investigation, they repeated the process using much larger (750 mL capacity) VHCs. As expected, they confirmed a substantial reduction in oropharyngeal deposition in adults inhaling the medication without a VHC, compared with the original CFC-formulated suspension product, a phenomenon also seen in earlier studies.(59,68) Where their patients used the smaller (149 mL) VHC, lung deposition with the pMDI alone was estimated to be 51±12% label claim (LC) mass/actuation. Importantly, lung deposition was not significantly increased by the use of this VHC either without delay (53±3% LC) or with a 2-sec delay (47±1% LC) between inhaler actuation and the onset of inhalation. Leach and Colice also reported 45±5% LC with no delay and 38±10% LC with a 2-sec delay when their large-volume VHC was used. The implication from these findings is that VHC capacity larger than about 150 mL may have little or no influence on BE assessments for this type of OIP. However, as an important caveat, it should be appreciated that the lack of a statistically significant difference in lung deposition via small- and large-volume VHCs may have been due to the small numbers of subjects tested. Nevertheless, it is also notable that their observation supports the same conclusion arrived at earlier by Dolovich that emitted aerosol-size characteristics (i.e., MMAD, fine particle mass, total mass) do not change much when the volume is greater than about 150 mL. This finding comes from a systematic assessment of cylindrical VHC geometry (length, diameter, and volume) on emitted mass and aerosol APSD characteristics from CFC-fluorescein placebo pMDIs (Fig. 8),(45) suggesting wider applicability beyond HFA-based solution formulation OIPs.
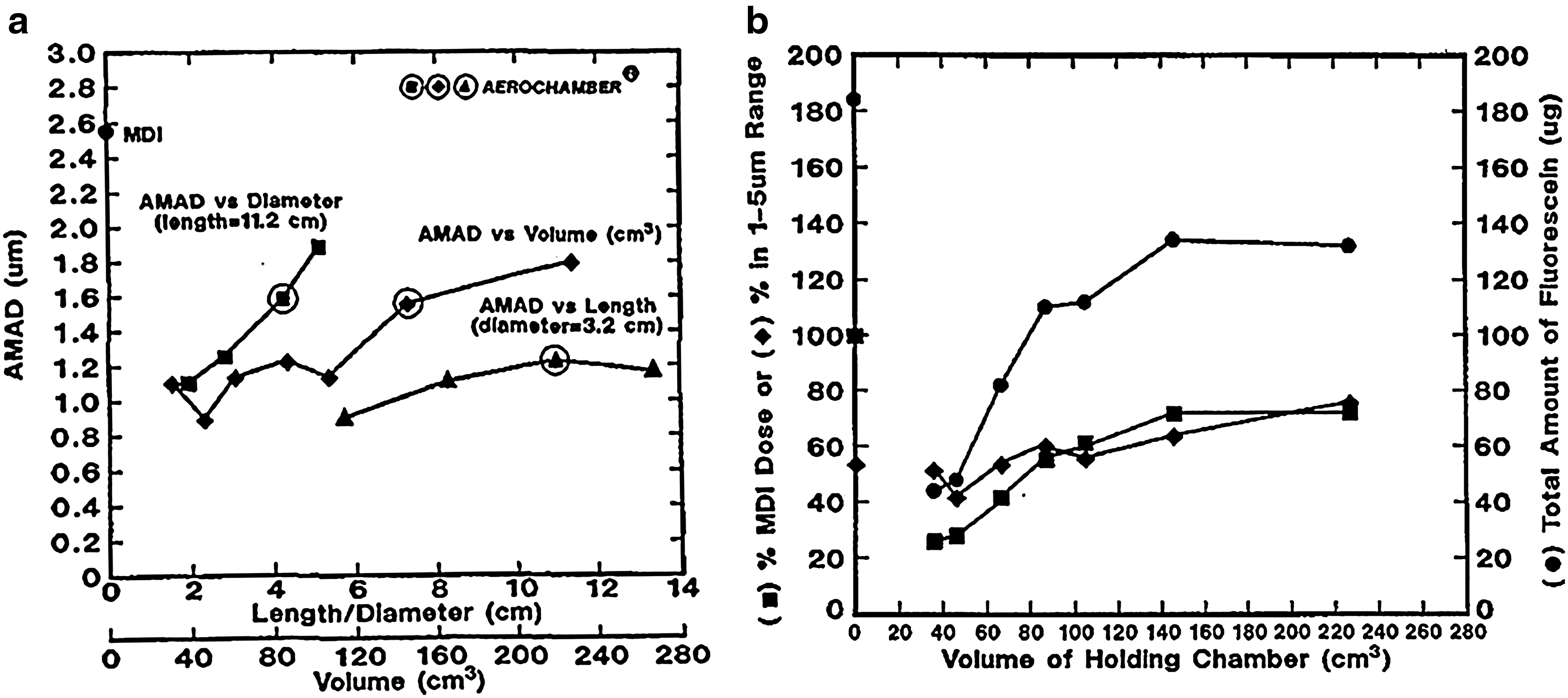
Effect of critical cylindrical-shaped VHC internal dimensions on aerosol characteristics assessed using fluorescent particles delivered by placebo pMDI [activity median aerodynamic diameter (AMAD)=2.6 μm].
Topical deposition from pMDI with VHC
Topical lung deposition is recognized as the measure from gamma-scintigraphy that is most likely linked to FPM.(21) Returning to the scintigraphic investigation by Leach and Colice,(67) it is interesting that the reduced variability they observed in their measures of lung deposition in instances where a VHC was present, may be interpreted as being indicative of improved consistency in dose delivery under such circumstances. Furthermore, the similarity in spatial deposition of inhaled particles in the airways of the lungs of their adult patients inhaling either with or without a delay, is consistent with the comparability of the laboratory-measured APSDs in terms of FPF<4.7μm (calculated from the ratio: FPM<4.7μm/TM) for this aerosol ex VHC, also with and without delay, observed in the Mitchell et al. study.(63) However, it has to be acknowledged that small differences in FPM<4.7μm, typically <20% LC mass/actuation, which are observed in such in vitro laboratory measurements, would be difficult to differentiate in regional deposition images due to the resolution of most gamma cameras.(46) In the case of Qvar, the more direct clinical measure of absorbed drug mass may be obtained from pharmacokinetic measurements, which may be more sensitive to changes in FPM of the inhaled BDP than gamma-scintigraphy. However, as the blood levels reflect total circulating mass of API, a significant change in regional deposition would likely be needed to modulate the resulting time–activity curve.(69) Even then, such serum-based measurements of API concentration would in turn need to be related to clinical markers appropriate to the inhaled glucocorticoid therapeutic class in order to complete the bridge between in vitro and in vivo measures of efficacy.
The laboratory investigation reported by Rahmatalla et al.,(70) undertaken at different simulations of inhalation flow rate via an idealized model of the human upper airway developed by Stapleton and colleagues at the University of Alberta, Canada,(71) also provides support for the finding that only small changes are associated with lung deposition by the addition of a 149-mL VHC when used with Qvar. The increase they observed in filter deposition (modeling total lung delivery) was not statistically significant (p<0.05) for flow rates of 28.3 L/min (42.2±3.9% LC without VHC; 44.6±4.1% LC with VHC) and 60 L/min (49.0±3.9% LC without VHC; 54.0±4.3% LC with VHC), only becoming statistically significant at 90 L/min [61.0±4.2% LC without VHC; 69.2±2.4% LC with VHC (p<0.005)], the highest flow rate they investigated. These findings from a model that is much closer to reality, as far as the geometry of the upper airway is concerned, even though it does not estimate the exhaled portion of the dose, strengthen the argument that any VHC-related effect on lung deposition with this formulation may be clinically insignificant at the average inspiratory flow rates achieved during use of these add-on devices that are normally close to 30 L/min.(72,73)
In contrast with solution formulations, many suspension-based products, such as salbutamol, were reformulated from CFC to HFA propellant with the purpose of retaining the original APSD profile of the emitted aerosol ex inhaler alone.(58) Thus, these OIP aerosols typically have MMAD values ranging from 2 to 5 μm, depending on API strength.(58,59,74,75) It is therefore to be expected that such aerosols will not penetrate the lungs as deeply as the finer-particle aerosols from HFA-solution formulations.(68) However, when a VHC is present, differences in predicted lung deposition as a function of VHC capacity may be reduced compared with the situation in which these pMDIs are used alone, based on in vitro data from at least one group (Cheng et al.(74)), albeit without breath simulation. This group used a hollow adult human airway replica “inhaling” at a constant flow rate of 30 L/min through either a 149-mL or 204-mL small-volume VHC. They reported “lung” deposition values for a suspension pMDI (HFA salbutamol having an MMAD of 2.21 μm, based on the impactor-sized portion of the emitted mass from this inhaler) in the range from 41.6% to 49.0% of the mass emitted from the pMDI actuator. This range of values may be compared directly with filter deposition as an estimate of likely lung deposition of 44.6±4.1% with the finer particles of Qvar (MMAD ca. 1.1 μm) from the previously described study of Rahmatella et al. at a similar (28.3 L/min) constant inhalation flow rate.(70)
Relationships between clinical- and laboratory-based measures of efficacy when a VHC is present
To complete the bridge between the in vitro data already discussed and clinical reality, it is clear that clinical studies focused on direct markers of bioavailability need to be performed that are demonstrated to be linked meaningfully to clinical efficacy. Such investigations therefore move well beyond imaging studies focused on the intermediate measurement of lung deposition. Nevertheless, it is important that feedback from the clinical outcomes reflecting changes in the delivery efficiency of the API is used to help drive the development of more clinically relevant in vitro test procedures for these devices. When a VHC is used to deliver a pMDI-generated aerosol, the near-elimination of deposition of API in the coarser mass fraction that would otherwise deposit to the oropharynx and larynx reduces the potential for subsequent systemic absorption via the gastrointestinal route.(45,75) However, systemic absorption to the pulmonary circulation also takes place via the extra-fine particles that deposit both in the distal airways and in the alveoli. The bronchial circulation may be another route for absorption and transport to the distal airways,(76) but will not be considered in this article. As a consequence, pharmacokinetic studies comparing relative performance of a particular formulation-pMDI combination used with and without a VHC need to be carefully designed to take into account modifications to both fine and coarse tails of the size distribution of the inhaled aerosol.(77) Examples of how this goal may be achieved are:
1. to take advantage of early lung-blood absorption compared with gastrointestinal tract-blood absorption of API, for both inhaled bronchodilators and corticosteroids(78);
2. to use the charcoal-block method to prevent gastrointestinal tract uptake from the swallowed oropharyngeal dose.(79,80)
Assuming that an appropriate methodology has been established to conduct a pharmacokinetic study to assess the clinical performance for a new entry pMDI with and without a VHC, two questions arise:
1. What are the accepted minimum differences in the metrics [area under the curve (AUC), the time (tmax) at which maximum API concentration (Cmax) is reached] that describe the time-dependent variation in drug concentration used to assess BE, and possibly also that may differentiate central versus peripheral absorption? These limits are key in justifying a claim that the drug product delivered through the VHC is clinically the same as from the pMDI alone.
2. How sensitive or specific do pharmacokinetic metrics have to be to relate meaningfully to in vitro APSD measurements? That is, can the AUC be related in a meaningful way to a laboratory-observed change in (a) the amplitude of the differential mass-weighted APSD and/or (b) its displacement to finer or coarser particle sizes?
The answer to the first question is related to the therapeutic class of drug(s) represented by the API(s) associated with the inhaler and the patient subpopulation evaluated (pediatric, adult, etc.), and therefore has to be answered on a product-by-product (81) and subpopulation-by-subpopulation basis.(82) It is therefore logical to set similar criteria established for BE in comparisons between test and reference pMDIs for the different therapeutic classes of medication to assessments comparing pMDI with and without VHC. Furthermore, these criteria should also be applied to comparisons between different VHCs used with the same pMDI-delivered drug product (Fig. 2).
In terms of responding to the second question, the ideal situation would be the establishment of in vitro–in vivo correlations (IVIVCs) that are well-defined mathematically for each class of therapeutic action (e.g., short-term β-2 agonist, long-term β-2 agonist, glucocorticosteroid). However, in practice, it has proven difficult to obtain such relationships even for the pMDI alone, without the added complexity of a VHC.(83) Furthermore, there is evidence from dry powder inhaler testing that the precision associated with the determination of in vitro measures is likely considerably narrower than the intrinsic variability of in vivo data.(80,84) In particular, the influence of differences in airway caliber from patient to patient can influence the variability in outcomes associated with clinical trials, even when care is taken to stratify patients by age, gender, disease stage, etc.(85)
Interestingly, Ehtezazi et al., working with several adult oropharyngeal models having well-defined differences in internal geometry, have hypothesized that the addition of a large-volume VHC to an HFA formulation may result in a reduction in the influence of natural variability in upper airway anatomy on lung deposition compared with that for the pMDI used alone.(85) This finding contrasts with that from an earlier similar study by the same group with an HFA-suspension formulation delivering a larger proportion of the aerosol mass in micron-sized particles (MMADs >2.4 μm) that indicated that variability introduced from differences in oropharyngeal geometry may not be reduced as much by the addition of a VHC.(86) Speculation as to the reasons for this finding could be that a greater proportion of larger particles generated from the suspension-formulated pMDI penetrated their model inlets via the VHC, and the variability associated with the deposition of such particles due to turbulence and inertial effects was greater than that associated with the finer particles from the solution-formulated product. The diverse findings between the two studies may therefore not be inconsistent with each other. In another investigation, Dolovich and Rhem, testing pMDI-delivered CFC-salbutamol with different induction port inlets connected to the ACI, found significant differences in the amount of API sampled by the inlets and the impactor as well as fine-particle fractions.(87) The former did not correlate with inlet volume, but did with the linear distance from the inlet opening to the inlet back wall (r2=0.763). Thus, the use of a standardized inlet, more closely resembling the human anatomy, may reduce the variability in the in vitro measurements as well as improve the correlation with clinical outcomes for pMDI-delivered products used with and without a VHC.
II. Current European and Canadian Regulatory Practice Regarding VHCs
The European Medicines Agency (EMA) guidance issued in 2009 set out the requirements concerning clinical documentation for OIPs for use in the treatment of asthma and COPD in adults, and in children and adolescents with asthma. In the context of S/VHCs, it indicates that a new OIP “can be authorized for use only if used with specific named spacer(s).”(1) Dissayanake, representing the UK Medicines and Healthcare Products Regulatory Agency (MHRA), in 2010 provided interpretation of the EMA guidance in the context of regulatory approval of a generic pMDI-based product as follows.(88):
“.…given that a generic and reference pMDI should be interchangeable and given the importance of spacer [VHC] use particularly in children, the failure to provide either in vitro or in vivo spacer [VHC] data confirming equivalence would generally preclude regulatory approval of the generic product.”
A draft guidance issued by Health Canada (HC) in 2007,(2) covering the subsequent entry of inhaled corticosteroid (ICS) products into the marketplace, also supports the testing of VHCs, through its recommendation:
“Since the drugs (ICS) are used in adults and children, evidence of compatibility with existing spacer devices or chambers is required for both adults and children.”
It must, however, be acknowledged that this draft guidance had very recently been revised in a version sent for public comment with a deadline of November 18, 2011, with the text above removed.(89) However, the original text has now been re-instated(89) following the March 2012 Health Canada Advisory Board meeting.
Uniqueness of each pMDI-VHC combination
It is less clear from the foregoing regulatory documents whether the “evidence” is to come from either laboratory or clinical testing or from both. However, it is likely that the substantial changes to the aerosol APSD when an S/VHC is present has been the driver underlying the recommendations. Furthermore, it is well known that the fine particle mass/actuation of medication delivered by a given VHC is dependent on the particular drug product under consideration, a phenomenon reported in laboratory studies undertaken in the late 1990s by Barry and O'Callaghan, comparing delivery of the same API (salbutamol) from two differently formulated products [Ventolin (GSK, plc) and Airomir (3M Pharmaceuticals)].(90) Similar observations were made by Dubus et al., comparing the in vitro performance of three different CFC-formulated salbutamol pMDIs with an HFA-formulated product of the same API with a variety of VHCs commonly used at the time in Europe.(91) It is also well established from in vitro studies that a specific VHC may perform differently with different APIs.(63,91–93) Similarly, a specific API in a specific pMDI may perform differently in terms of emitted fine particle mass/actuation (FPM), if inhaled through different VHCs.(90) From this evidence supporting the uniqueness of each type of VHC in its interaction with the currently available range of pMDI-delivered products, it is highly probable that substitution of one type of VHC with another without the support of clinically relevant in vitro testing, coupled possibly by clinical assessment, will be inadequate for regulatory approval.
The importance of VHC design
Both TM and FPM vary with VHC geometry, as has been previously discussed in relation to aerosol depletion processes. Other physical properties, such as inhalation valve design and the presence of surface electrostatic charge, if not eliminated,(94) are additional critical factors affecting performance, in addition to the specific inhalation variables associated with use.(95–97) Thus, it follows that the distribution of deposited aerosol in the lung and consequent clinical response to the API(s) present cannot necessarily be assumed to be equivalent if either a different VHC is used with the same drug product, or if a different formulation of the same API is used with the same VHC. Given these uncertainties, which have not yet been resolved unambiguously,(83) the EMA has therefore opted for the cautious approach already described. Importantly, their guidance goes further by acknowledging that the add-on device has to be appropriate for the intended patient population (infant, small child, adolescent, adult).(1)
Flow-dependent properties of the aerosol, cascade impactor operating flow rate, and metrics associated with aerosol APSD
The EMA guidance further states that knowledge of in vitro behavior, in particular the flow-dependent properties of the aerosol available to be inhaled, is important in addition to clinical performance for establishing both clinical efficacy and safety of the product administered via the pMDI-VHC combination for a new or subsequent market entry product.(1) It follows that a systematic examination of inhalation flow-rate in vitro characteristics through laboratory-determined measures of performance would provide an important data set to support the clinical development program.
Measurements of TM by filter collection at the patient interface of the VHC using a breathing simulator to create age-appropriate respiratory patterns is relatively easy to do, and approaches to validate this type of study have been reviewed recently by Schultz et al.(98) Added realism can be achieved by replacing the breathing simulator by patients or volunteers who inhale from the VHC via a filter located in a suitable housing around which their lips seal.(98,99) This type of study has the advantage that natural variability in breathing patterns can be incorporated into the study design, a feature that, though possible with a breathing simulator, is time-consuming to execute, relying on a priori knowledge of the range of breathing patterns that are pertinent. However, particle-size distribution information cannot be obtained in this way.
Investigations to determine the flow-rate dependency of APSD are relatively easy to undertake, but require a cascade impactor. As an example of what can be done, Mitchell et al. reported measurements of the APSD of pMDI-delivered HFA-fluticasone propionate undertaken by operating an ACI at different flow rates within its operating range (28.3, 45, and 60 L/min).(100) Interestingly, the resulting APSDs obtained with the pMDI alone (HFA-fluticasone propionate; 125 μg/actuation) were not found to be very flow rate-dependent in this study (Fig. 9), a slight displacement to finer sizes only becoming evident at the highest flow rate. FPM<4.7μm increased slightly, but significantly, between 28.3 L/min and 45 L/min with both groups of large- and small-volume VHCs that were evaluated (Fig. 10). However, a further increase to 60 L/min did not result in a corresponding gain in output from either group of devices. A similar approach should be possible using the NGI within its wide operating flow-rate range from 15 L/min to 100 L/min.(33,101) In principle, it should also be possible to switch to an alternative impactor, such as the low-flow Marple-Miller (MMI) design [model 150P, MSP Corp., St. Paul, MN (Fig. 11)], if it becomes necessary to obtain APSDs at flow rates lower than the normal range for either the ACI or NGI, as the low-flow MMI is designed to operate at both 4.9 and 12.0 L/min with the cut-point size of 4.7 μm for FPF matching that of the ACI at 28.3 L/min.(102)
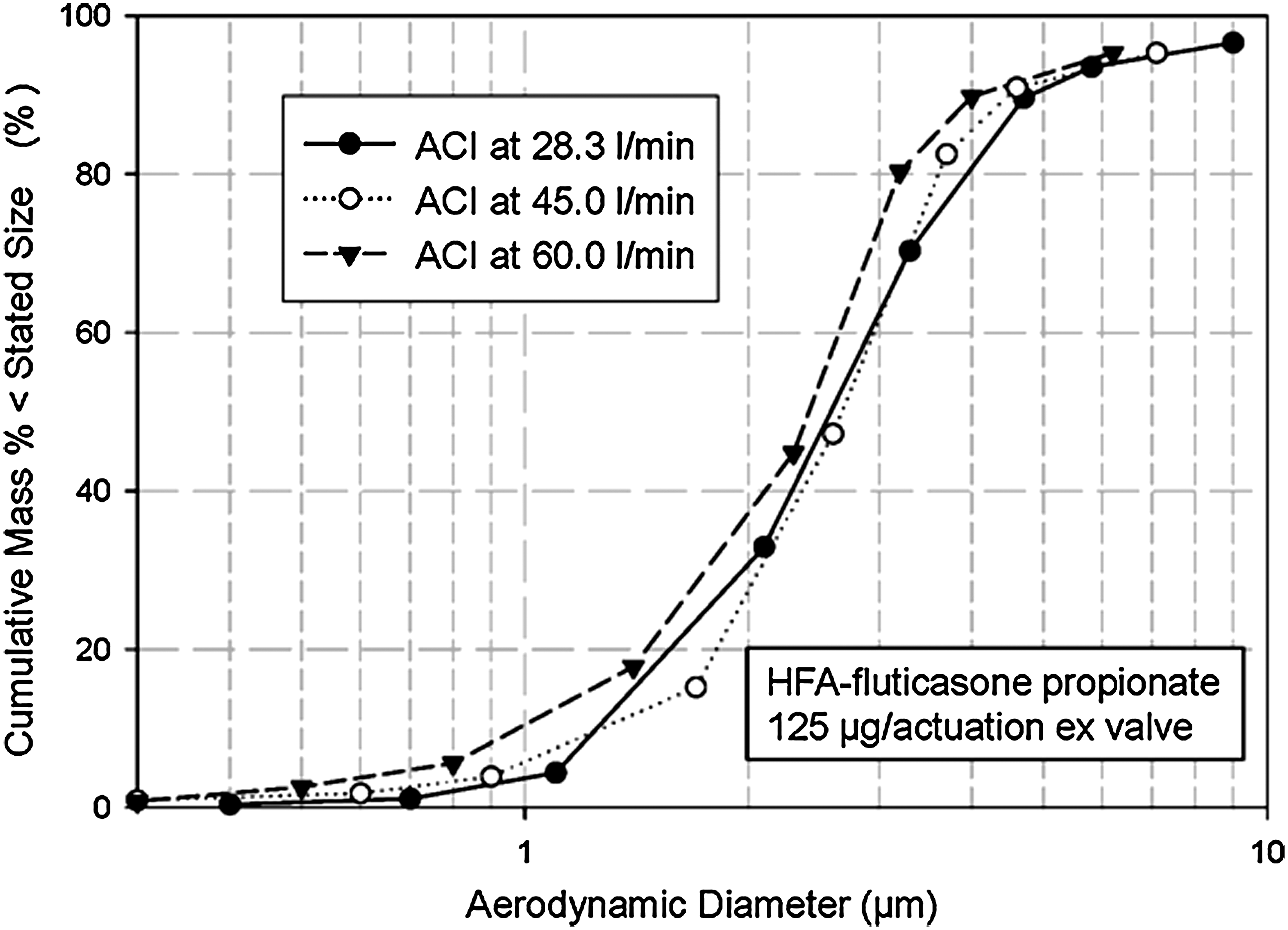
ACI-measured APSD scaled to IM at three different flow rates for HFA-fluticasone propionate showing only slight displacement to finer sizes as flow rate was increased; cascade impactor effective cutoff diameters change to match sampling flow rates; the mass retained by the USP/Ph.Eur. induction port was close to 4 μg at each flow rate.
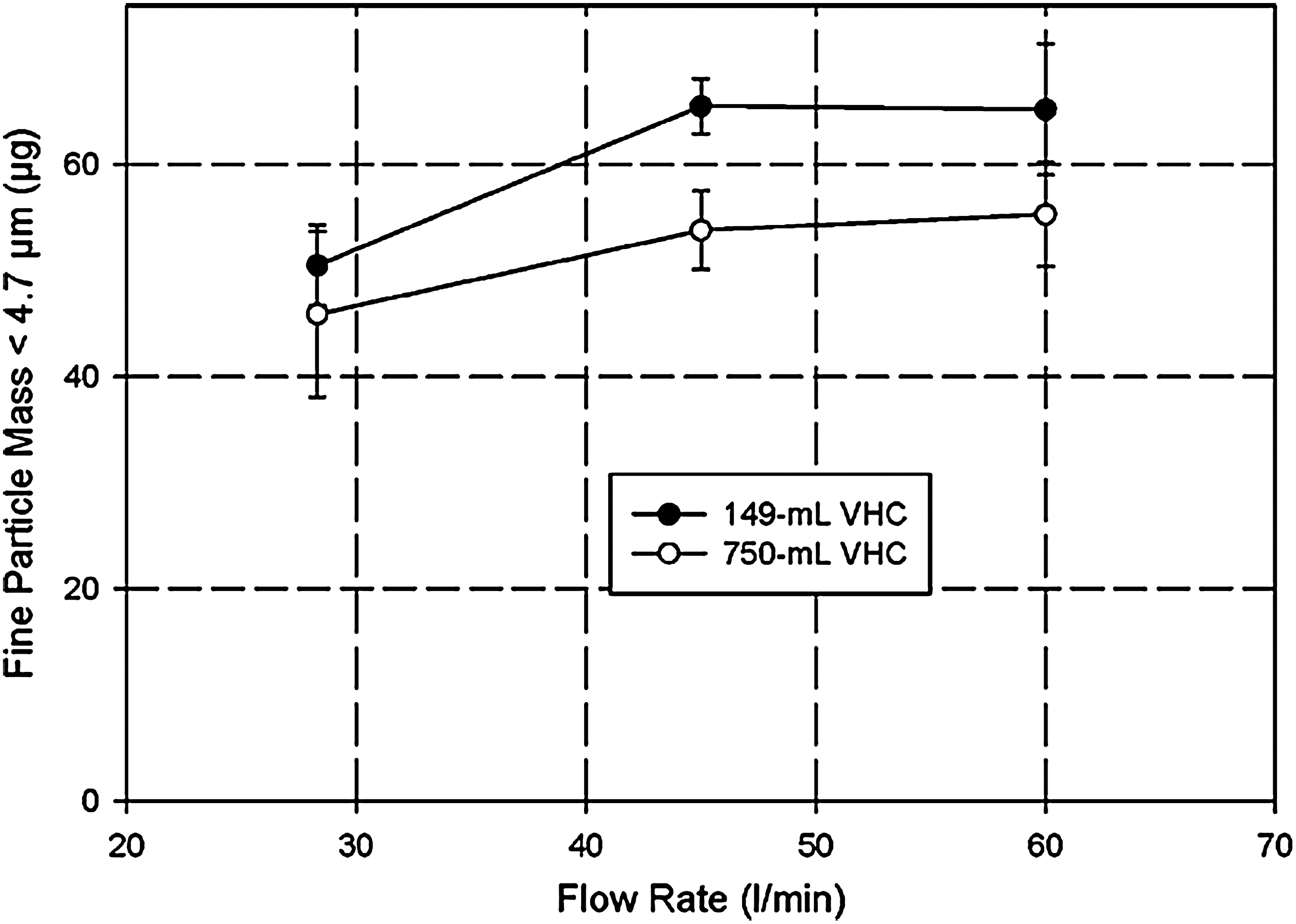
Fine particle dose (mass) <4.7 μm aerodynamic diameter of aerosols from large- and small-volume VHCs tested with HFA-fluticasone propionate (125 μg/actuation) as a function of flow rate. FPM<4.7μm increases only slightly at the higher flow rate.

Low-flow Marple-Miller 5-stage cascade impactor developed for testing VHCs intended for use by infants or small children.
Measures of therapeutically important subfractions, such as FPM and EPM (for cases where the MMAD of the emitted aerosol ex pMDI is <ca. 2.0 μm), as well as the full APSD, are also likely to be needed as key components of the in vitro data set. If EPM is reported in addition to FPM (Fig. 4), it makes sense to structure the data such that the lower size boundary of FPM is defined contiguous with the upper size limit for EPM. The FDA recognizes this approach in their draft CMC guidance for MDI and DPI products, where it is stated:
“acceptance criteria may be proposed in terms of appropriate groupings of stages and/or accessories.”(14)
The equivalent HC-EMA harmonized guidance refers to the same process as stage pooling.(27,28)
Unfortunately, the important question—whether such a study should be confined to the determination of APSD by cascade impactor at several different but fixed flow rates, or extended to the measurement of delivery performance simulating patient breathing—is left moot.
When considering the most appropriate measurement approach for APSD in laboratory-based testing, it is important to note that cascade impactors are intended to be used only at fixed flow rates, because, by design, their stage effective cutoff diameters are flow rate-dependent.(23,103) Hence, testing at various fixed inhalation flow rates that are physiologically relevant to the intended patient age group (i.e., time-averaged and peak inspiratory flow rates) by this method would ideally be needed to characterize fully how the aerosol APSD characteristics are influenced by the continuously varying flow-time pattern associated with the inspiratory portion of a representative breathing pattern. Although several attempts have been made to operate cascade impactors in conjunction with breathing simulators to determine time-averaged APSDs from simulations of tidal breathing,(104–106) such arrangements are complex. In particular, there is the risk that the aerosol emitted by the inhaler may be improperly sampled (i.e., anisokinetic sampling of the breathing simulator-created variable flow rate-time profile from the inhaler to maintain the constant flow rate required by the impactor). VHC valve function may be compromised if pressure pulsations are present in association with the supply of makeup air for the impactor during the exhalation portion of each breathing cycle.(25) At the present time, validation of a breathing simulator-cascade impactor system with one or more aerosols having independent and well-defined APSDs, has not been provided as a demonstration of the accuracy of the CI measurements. Ideally, such a validation should be undertaken with a range of well-defined breathing patterns pertinent to the major age categories, such as those provided in the Canadian standard for S/VHCs.(107) Until such a study is successfully undertaken, it is improbable that the pharmacopeial authorities will recognize such equipment as being within their scope for the in vitro assessment of VHCs, and we support this position.
Pharmaceutical Quality of Inhalation Products: HC and EMA Positions
A major step forward in defining the nature of in vitro test requirements for OIP performance characterization in the context of quality control was taken in 2006 by the simultaneous publication of mostly harmonized guidance documents by the EMA(27) and HC.(28) However, it is important to note that Appendix 3 of the EMA text included an important exception to full harmonization, and it affects how VHCs are to be considered. Here, the EMA stipulated the following four items to be considered in submissions for approval:
“When a spacer or holding chamber is required for administration of the product to a particular patient population (e.g., paediatrics, administration of high dose steroids), its use should be validated. Relevant information on the spacer/holding chamber must be given in the Summary of Product Characteristics,” and “In addition to in vitro studies, the suitability of the spacer should be supported by appropriately designed clinical studies. Any claims exceeding instructions for use and handling, e.g., reduction in the amount of large particles, must be supported by in vitro data.”(27)
As might be expected from regulatory advice focusing on pharmaceutical quality, the type of laboratory testing for VHCs outlined in the harmonized EMA-HC guidance documents is assumed to be taken from the pharmacopeial sources, and therefore reliant on the methodologies provided in Monograph 2.9.18 of the Ph.Eur.(34) and Chapter 601 of the USP(35) for the determination of aerosol APSD. It is, however, important to realize that although these pharmacopeial procedures are fully appropriate to fulfill the requirements of these product quality guidelines, they were not developed specifically for assessing clinical efficacy and safety. In particular, the methods involve great simplification of the inhalation process, so their value in considerations of BE is questionable.(21)
An obvious example of the simplification from clinical reality is the use of the Ph.Eur./USP inlet that comprises a length of stainless steel tubing with specified entry and exit dimensions, incorporating a well-defined, right-angle bend between the two (Fig. 12), rather than an anatomically correct upper airway for the induction port that is intended to represent the oropharynx.(108) Improvements to more closely mimic reality by means of a designed-for-manufacture inlet having comparable particle-capture characteristics to an anatomically correct upper airway are in progress, most notably the work of Finlay and colleagues with the development of the “Alberta” idealized inlet (AIT).(109,110) A commercially available version of this model inlet has recently become available (Fig. 12),(111) but a significant body of validation data will be needed before the compendial authorities are likely to consider adopting it. However, early signs are promising, with APSDs from a pMDI-delivered salbutamol measured by this inlet using the NGI showing less broadening as well as displacement to finer particle sizes associated with increased deposition of the coarse fraction in the AIT, in direct comparison with similar data determined by a Ph.Eur./USP induction port.(111) Similar behavior has also been observed when the NGI is replaced with the ACI.(112) Currently, only the adult version of the AIT is available. A child version is in late-stage commercialization. This leaves an ongoing need for a corresponding idealized inlet mimicking the infant upper airway, which is markedly different from that of adults.(113)

Open “Alberta” idealized adult throat inlet for more clinically appropriate measurements of OIP APSDs (left) compared with USP/Ph.Eur. induction port (right).
A further oversimplification is the subjection of the VHC being tested to a steady air flow originating from the vacuum pump used to operate the CI. This arrangement fulfills the requirements of the compendial methodologies for pMDI quality assurance, ensuring that the cascade impactor is ideally operated at a fixed flow rate only. However, under these circumstances, it is important to note that proper functioning of the inhalation valve and exhalation valve (if equipped) of the VHC is not evaluated. The dependence on fixed flow rate sampling from the pMDI+VHC system could therefore result in acceptance of devices with valves that might not operate properly, a situation that has been observed in one in vitro study undertaken at the low flow rates associated with infants and small children.(114) However, it is important to note that the EMA guidance also suggests that the fine particle mass protocol used for routine testing of an inhaler can be altered to permit simulation of more realistic patient use (e.g., tidal breathing),(27) so there appears to be scope to submit data obtained under such more clinically relevant conditions. Performance evaluation of a pMDI+VHC simulating respiration cycles should be performed with the breathing pattern appropriate to the intended patient age group. Thus, in vitro data acquired for a VHC intended for infant or small-child use with an age-appropriate breathing pattern would likely be of more value in considerations of BE than an attempt to extrapolate likely pediatric performance from measurements obtained with an adult breathing pattern.
III. Laboratory Evaluation of VHCs to Provide Meaningful In Vitro Data in the Context of Establishing Bioequivalence
Although the European regulatory recommendations concerning clinical documentation recognize the need to evaluate add-on devices in ways that more closely mimic clinical use, rather than by means that simply assess product quality,(1) the present reality is that there is little in the way of assistance in the regulatory, compendial, or standards literature to help those tasked with the laboratory evaluation of these devices in clinically appropriate ways. In the process of designing a regime for more clinically relevant laboratory testing, it should be recognized at the outset that, for a variety of reasons (patient demographics, anatomic variability, presence of comorbidities, etc.), the intrinsic variability associated with well executed in vitro measurements is likely to be substantially less than the typical variability of clinical measures from pharmacokinetic studies to characterize BE.(83,84,115) This comparison is likely to be true even for the complex-to-execute methodologies involving cascade impaction.(24,116)
If it is accepted that in vitro testing will be required of at least one design of VHC to meet European and Canadian regulatory requirements for new pMDI product approvals, the question: “How should VHCs be evaluated in the laboratory” requires an answer that involves establishing a common understanding among stakeholders of the minimum requirements needed to be met to provide metrics that parallel clinical performance and ultimately BE. In this context, there is currently an ongoing dialogue between regulators and experts within the inhaled pharmaceutical product community that will need to be monitored to determine what additional requirements to the existing array of laboratory measurement techniques for OIPs are being proposed.(11,12,117)
The Canadian Standard on Spacers and VHCs: Clinically Relevant Laboratory Test Methods
Delayed sampling from the VHC
About 10 years ago, the lack of clinically pertinent laboratory test procedures for S/VHCs was partly addressed by a group of Canadian stakeholders involving clinicians, pharmacists, pharmaceutical companies marketing pMDI products, manufacturers of S/VHC products, academia, and HC. The result was a Canadian Standard, which for the first time set out detailed methods for the evaluation of these add-on devices, including sections on the simulation of delayed inhalation following inhaler actuation.(107) At the outset, it was understood that EPM, FPM, and CPM were of key importance for assessing or predicting likely clinical performance between the pMDI alone and pMDI with S/VHC products.(118,119) It was also recognized that, as a start, the in vitro performance for the pMDI alone should be assessed by determining the APSD of the emitted aerosol, and obtaining the resulting subfractions corresponding to EPM, FPM, and CPM, with no delay interposed between inhaler actuation and the onset of sampling. This decision was made for the practical reason that once the pMDI is actuated, the aerosol plume rapidly disperses unless inhaled immediately using an optimal technique such as the “closed lips” or “open mouth” methods.(120,121) However, when a pMDI+VHC combination is to be evaluated, the consensus was that comparisons between VHCs would be better based on measurements with delay rather than with no delay, given the high probability that patients using these add-on devices will have less than perfect coordination.(122,123) The in vitro testing component in the Canadian standard was therefore structured to incorporate benchmark testing of VHCs with no delay for comparisons in relation to the performance of the pMDI alone. Such evaluations, mimicking perfect patient inhaler technique, were augmented by evaluation of VHC performance with the preferred delay interval set at 2 sec, as a good approximation of a poorly coordinated user(124,125) (Table 2). The apparatus illustrated in Figure 11 was constructed at Trudell Medical International to assist with cascade impactor-based measurements of APSD of the aerosol ex VHC with delay.(53) The mouthpiece of the VHC is coupled to an adapter that in turn fits to the entry of the USP/Ph.Eur. induction port. A vertically mounted shutter locates on a solenoid-operated ledge at the base of the adapter assembly. In this position, the exit from the VHC is closed, but at the same time impactor is allowed to sample ambient air normally via a slot machined on the side facing the induction port. A microphone detects actuation of the pMDI. After the programmed delay interval has elapsed, the solenoid is actuated, retracting the ledge and causing the shutter to drop by gravity into the lower position in which an opening aligns with the VHC mouthpiece, thereby permitting an almost instantaneous increase in flow from zero to the desired flow rate.
Inspiratory flow rate as a test variable
The important issue of choosing a test flow rate appropriate to the intended patient use was also accommodated in the section concerned with measurement of aerosol APSD. The option was provided to use a suitable cascade impactor operating at a lower flow rate than the standard 28.3 L/min for the ACI or 30 L/min for the NGI, commonly used to test products intended for adult use. The low-flow MMI (Fig. 11) has been used for the evaluation of two small-volume VHCs at both 4.9 and 12 L/min, based on measures of TM, FPM<4.7μm, and FPF<4.7μm (Table 4).(126) However, the acquisition of benchmark data for the pMDI alone at the lower flow rate was found to be complicated by the fact that the volume increase associated with plume expansion temporarily pressurized the sampling system to the CI, resulting in loss of part of the aerosol through the air-intake vents that form part of the mouthpiece-actuator assembly. In principle, the use of the more recently developed NGI at 15 L/min, the lowest flow rate for which calibration data are available,(101) should also be suitable for testing VHCs intended for use by small children. However, validation studies confirming the appropriateness of this particular impactor for this role have yet to be published.
Data are from Ref. 113.
Simulation of tidal breathing
The Canadian standard also introduced the simulation of tidal breathing in answer to the need to assess the pMDI-VHC as a complete system and provide more clinically relevant data than just sampling the emitted dose at constant flow rate.(23) Several well-defined breathing patterns were established covering the main categories of patient by age group (Table 5), and the rationales underlying these particular choices were explained in a supporting publication by Dolovich and Mitchell.(118) These breathing patterns were interpolated from the work of Stocks and Hislop published in 2002(113) and have subsequently been referenced by an International Standard for the design verification of OIPs, including add-on devices.(127) The Canadian standard requires that the total emitted mass/actuation be determined initially with inhaler actuation coincident with the onset of inhalation (TMcoordinated), mimicking the optimal condition of a fully coordinated user. Repeated testing is then proposed to be undertaken, this time simulating the worst-case condition in which inhaler actuation coincides with the onset of exhalation (TMuncoordinated).(118) The ratio of the two values is taken to be indicative of the overall performance of the VHC, because the assessed losses of medication inside the chamber are due to normal particle removal processes that occur once the aerosol plume has formed, as well as those arising possibly from imperfect valve function. Although no size fractionation has taken place, breath simulation evaluates the mechanical operation of the add-on device as it is likely to be used by the patient. The emitted fine particle mass of API per actuation fully coordinated (FPMcoordinated) or fully uncoordinated (FPMuncoordinated) can subsequently be obtained as the product of FPF (derived from the cascade impactor measurements in a parallel study) and TMcoordinated or TMuncoordinated, respectively.
Facemask interface: dead space and leakage as variables of importance
The proper evaluation of the VHC with facemask as patient interface poses some additional difficulties, as the compendial apparatuses, being focused on the assessment of OIP quality, were designed assuming that the patient interface would be a mouthpiece, which is the normal interface for OIPs. In particular, the correct realization of dead space within the facemask when in use(128,129) (Fig. 13), as well as the elimination of leakage at the facemask–face seal,(130) are paramount for accurate in vitro measurements to be made,(131) as well as in assessments of clinical efficacy.(132) When the original Canadian standard for spacers and holding chambers was created, there was recognition that the development of in vitro test methods had not yet progressed to the point at which a sufficiently robust apparatus for the purpose of testing VHC-facemask combinations could be recommended.(118) The compromise practice of removing the facemask and coupling the mask adapter, either directly to the induction port for impactor measurements or to a filter holder for breathing simulation testing, was therefore advocated as an interim measure. This concession recognized the fact that better methodologies were in the process of being developed, but were not yet sufficiently validated to become candidates as compendial apparatuses. Since then, several studies involving the use of model faces have been published.(133,134) These laboratory studies have been undertaken sometimes with rudimentary realizations of the upper airway, but in other instances the investigators have made use of anatomically correct oro- or nasopharyngeal geometries, such as the nasopharynx contained within the Sophia Anatomical Infant Nose-Throat (SAINT) model.(135) The increase in interest is indicative of the importance of the topic, and was reviewed by Mitchell in 2008,(136) finding at that time that the SAINT model was the most advanced if its kind. However, it was noted that it is necessary either to apply sealants or to compress the facemask beyond normal to eliminate leakage with the rigid facial structure that is incomplete above the bridge of the nose.
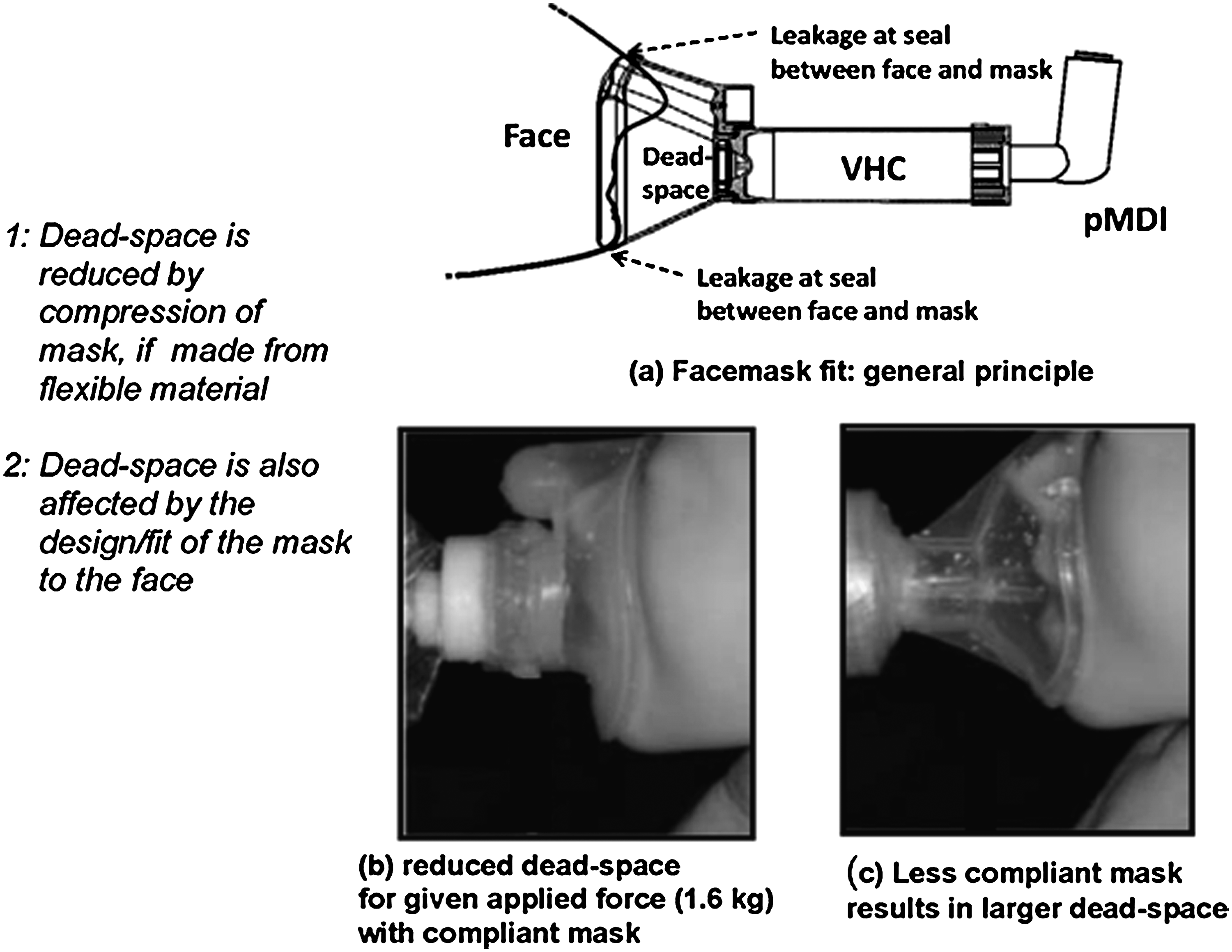
Schematic illustration of VHC-facemask interaction with face of user in the context of defining dead space and leakage avoidance.
In the 2008 revision of the Canadian standard, these advances were recognized in a new informative annex by the statement:
“If an exhalation valve is present in the mask, the testing of the S-HC should be carried out with the face mask in place.”(107)
However, this attempt at guidance toward clinically appropriate testing was qualified by the following statement:
“While manikins (mannequins) are available or can be built with realistic facial geometries and some test procedures are described in the literature, the methodology has not yet advanced to the point at which normative guidance on testing procedures can be provided in this Standard.”
The caveat recognized that apparatuses validated to compendial standards were still not available.
More recent developments, such as the ADAM-III infant face-nasopharyngeal model,(137) may provide a way forward toward developing suitable clinically realistic standard models. The ADAM-III approach combines an anatomically accurate upper airway with a close and reproducible facsimile of the mechanical resilience of the facial skin and underlying soft tissues coming into contact with the facemask (Fig. 14). This degree of realism has been found essential in order to arrive at the correct dead space between the inhalation valve of the VHC and the open mouth of the patient when the facemask is applied to the face model with a clinically appropriate force of 1.6 kg.(138) Both small-child and adult versions of this model are in the process of being developed in order to make the ADAM-III concept fully applicable to the range of patients for which VHCs are prescribed. In the context of developing methodologies that can be incorporated into the compendia, the ultimate goal should be that face models both are readily reproducible and have anatomically accurate face/upper airways standardized to simulate, as a minimum, infant, small-child, and adult patients. Ultimately, the critical dimensions and other pertinent physical characteristics of these models, such as “skin” responsiveness to applied pressure, should become established sufficiently well that they can be defined in quantitative terms. Under such circumstances, measures such as EPM, FPM, and CPM could be accurately determined for VHC-facemask combinations by the use of both cascade impaction and breath simulation in parallel measurements, as is currently possible for VHC-mouthpiece systems, by following the practices outlined in the Canadian standard.
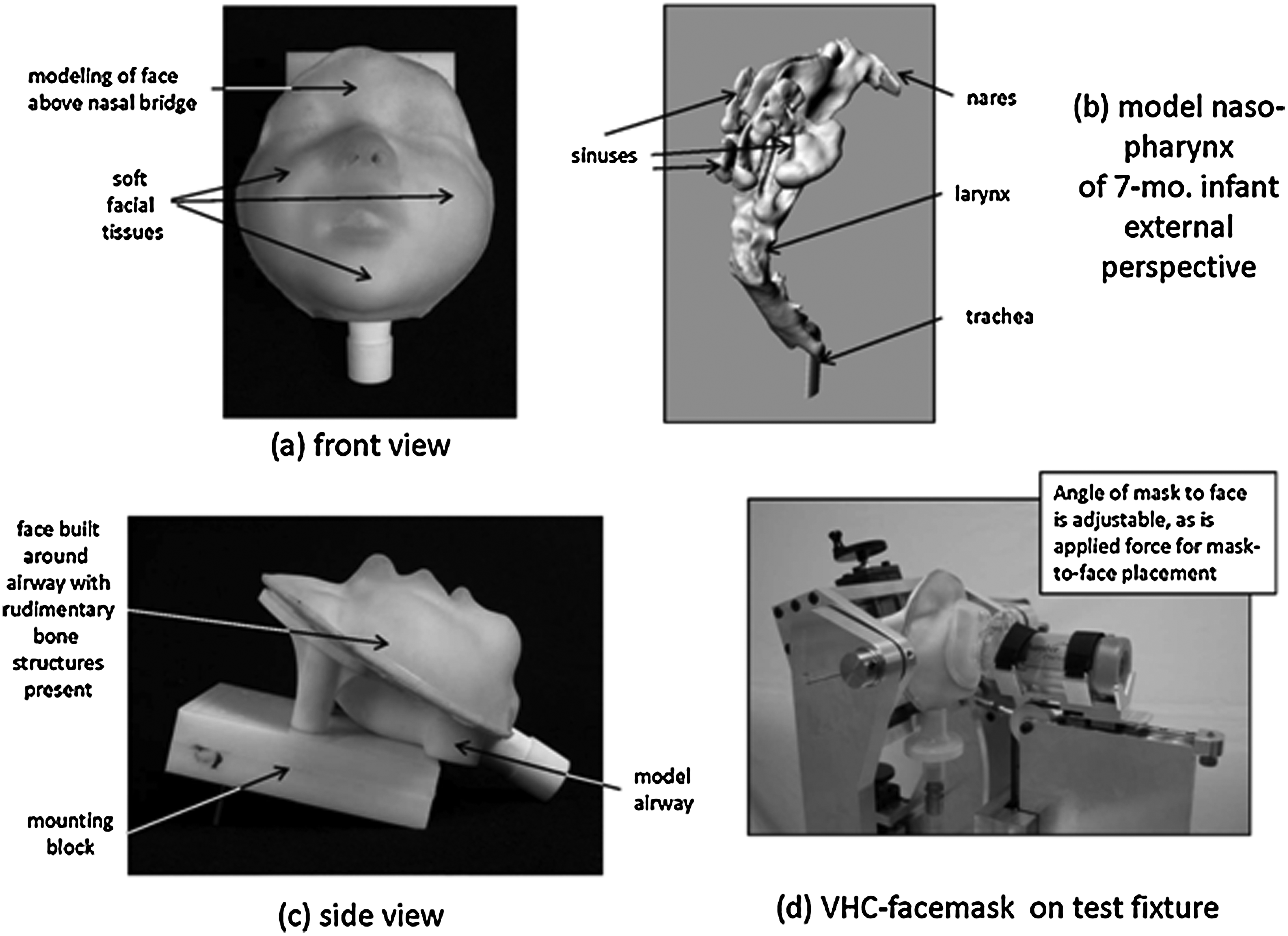
ADAM-III infant face model developed for the evaluation of VHC-facemask systems.
The in vitro testing component of the Canadian Standard has recently been recognized as having the potential to be adopted as an informative monograph in the USP. The pertinent Stimulus to Revision article, which is the first step in the process that could ultimately lead to the new chapter, has completed public review and comment.(139)
Other Considerations
Beyond the establishment of meaningful in vitro criteria with which to assess BE either for two inhaled drug products used with a nominated VHC or for two different VHC designs with the same pMDI-based product, there remains the important related but wider question of whether or not a marketed VHC is fit for purpose if a critical component, such as the inhalation valve, could become nonoperative under realistic conditions of use.(113) The possibility of pairing a poorly in vitro performing VHC to a given pMDI product raises concerns about potential safety as well as efficacy of the inhaled drug delivery system.
As has been previously identified, SMEPs as well as innovator products must first be evaluated by in vitro testing with and without a named VHC to be able to progress to the clinical platform for regulatory approval. Presumably, poor performance of the named VHC would become evident as such data are assessed either initially by the pharmaceutical company or later by the regulatory agency. However, the way the comparison between “test” and “reference” products is currently structured, the same poorly performing VHC used in a head-to-head clinical comparison of two pMDI-based drug products might well result in an outcome in which the two products are deemed equivalent, but with suboptimal in vitro performance and possibly clinical efficacy. The requirement that in vitro testing of VHCs be done in ways that challenge the effective operation of key components of the VHC should therefore be a consideration in future updates to the pertinent regulatory guidance documentation in all regions in which SMEP-to-innovator comparisons are likely to be submitted for approval.
Conclusions
Table 6 summarizes the main learning points arising from this article. In considerations of BE, the assessment of the in vitro performance of a given pMDI-based OIP in isolation from its likely prescribed use with a VHC fails to recognize the widespread recommendation, by both national and international clinical guidelines, that these add-on devices are necessary to achieve optimal therapy for patients of all ages with imperfect inhaler coordination for the management of bronchoconstrictive diseases such as asthma and COPD. Although the existing compendial procedures are appropriate to evaluate the performance of the pMDI alone, the evaluation of pMDI-VHC systems necessitates that testing take place in ways that more closely mimic actual patient use. At the same time, such test methodologies must ensure that key components of the add-on device are properly evaluated to demonstrate effective function. Given this situation, there is therefore an urgent need to support current regulatory practice in the EU and Canada by specifying in precise terms what is required to achieve an acceptable standard for clinically relevant laboratory testing of VHCs. The Canadian standard for spacers and VHCs provides a pathway toward achieving this goal. Going beyond this standard, the rapid development of widely available and age-appropriate standardized face models, preferably incorporating either an anatomically correct or possibly idealized oro- or nasopharyngeal airway, is needed so that effective aerosol delivery via the facemask interface can be verified. Such additions to the test equipment will likely be sufficient to complete the suite of in vitro testing that will be needed in support of BE comparisons in which a VHC is involved in the delivery of medication from the OIP.
Footnotes
Author Disclosure Statement
Myrna Dolovich has no conflicts of interest to declare. Jolyon Mitchell is a full-time employee of a manufacturer of VHCs mentioned in this article, but otherwise has no conflicts of interest.