
Abstract
Abstract
The purpose of this article is to document the discussions at the 2010 European Workshop on Equivalence Determinations for Orally Inhaled Drugs for Local Action, cohosted by the International Society for Aerosols in Medicine (ISAM) and the International Pharmaceutical Consortium on Regulation and Science (IPAC-RS). The article summarizes current regulatory approaches in Europe, the United States, and Canada, and presents points of consensus as well as ongoing debate in the four major areas: in vitro testing, pharmacokinetic and pharmacodynamic studies, and device similarity. Specific issues in need of further research and discussion are also identified.
Executive Summary
From the technical perspective, there is a general consistency among regions in the scope of important in vitro tests (e.g., dose uniformity, aerodynamic particle size distribution, sameness of active pharmaceutical ingredients (APIs), similarity of excipients and devices), although specific acceptance criteria—where they have been explicitly stated—vary.
Special challenges are posed by multiple-API products (sometimes called combination products or fixed-dose combination products) and by consideration of pediatric populations.
Introduction
Over the past few years, several workshops have discussed approaches for demonstrating (bio)equivalence of OIPs that would be needed either for marketing authorization of a generic/second-entry product, or for justifying changes/line extensions for a brand-name/innovator product. In 2009, the Product Quality Research Institute (PQRI) sponsored a workshop “Demonstrating Bioequivalence (BE) of Locally Acting Orally Inhaled Drug Products.”(1) In 2010, PQRI in coordination with the Respiratory Drug Delivery Conference sponsored a workshop “Role of Pharmacokinetics in Establishing Bioequivalence for Orally Inhaled Drug Products,”(2) focusing particularly on the potential use of pharmacokinetics (PK) as the sole indicator of in vivo BE for locally acting OIPs. Later in 2010, a “European Workshop on Equivalence Considerations for Orally Inhaled Products for Local Action” was coorganized by the International Pharmaceutical Aerosol Consortium on Regulation and Science (IPAC-RS) and the International Society for Aerosols in Medicine (ISAM). The objective of this European Workshop was to continue the constructive dialogue among industry, regulators, and academic researchers started at the preceding two workshops but to focus it on the European views regarding OIP equivalence determinations. This article summarizes the discussions at this ISAM/IPAC-RS European Workshop (“the Workshop”).
In this article, the term Test product (T) refers to the OIP for which equivalence needs to be demonstrated, for example, a generic product or a modified innovator's product. Analogously, a Reference product (R) refers to the OIP that serves as a basis for comparisons in equivalence determinations, that is, the original product.
This article reflects the current state of understanding and knowledge gaps in this field as revealed at the Workshop; it should not be viewed as regulatory advice, and it may not fully represent the positions of individual authors or of any organization with which they are affiliated.
Overview of Regulatory Requirements for Demonstrating Equivalence of OIPs for Local Action
The European regulatory perspective on demonstrating therapeutic equivalence of OIPs is described in a CHMP Guideline that was issued as final in early 2009 and came into force in August 2009.(3) This guideline applies to products that are used to treat asthma and chronic obstructive pulmonary dissease (COPD), and is relevant for a variety of OIP dosage forms, including pressurized and nonpressurized metered dose inhalers (MDIs), dry-powder inhalers (DPIs), and solutions and suspensions for nebulization. In Europe, locally acting products such as OIPs do not meet the strict definition of a “generic medicinal product.”(4) Such products are commonly called “hybrid medicinal products” and their submission basis is described in article 10.3 of directive EC/2001/83.(5)
Overall, the EMA guideline sets general EU expectations for approval based on equivalence studies. Regardless of the legal submission basis discussed above, individual Member States have a sovereign right on a scientific basis to object to marketing approval of a product that they associate with lack of proof of sufficient risk/benefit balance. In the European Mutual Recognition or decentralized procedures, these concerns and differences could be resolved through an arbitration process leading to a common decision. In national procedures, however, EU Member States may have different final requirements. Furthermore, interchangeability/substitution (which has implications for reimbursement) is always decided at the national level. This gives each country the flexibility to consider such factors as their population's needs, climate, historical data, etc.
As depicted in Figure 1, the European guideline describes a stepwise approach to demonstrating therapeutic equivalence using in vitro and in vivo data. In brief, the concept first uses in vitro comparisons, followed, if needed, by PK studies, and finally PD or clinical effect studies. If equivalence is demonstrated at any of these major steps, the subsequent studies are not required. Each step is discussed in more detail in the remainder of this article.
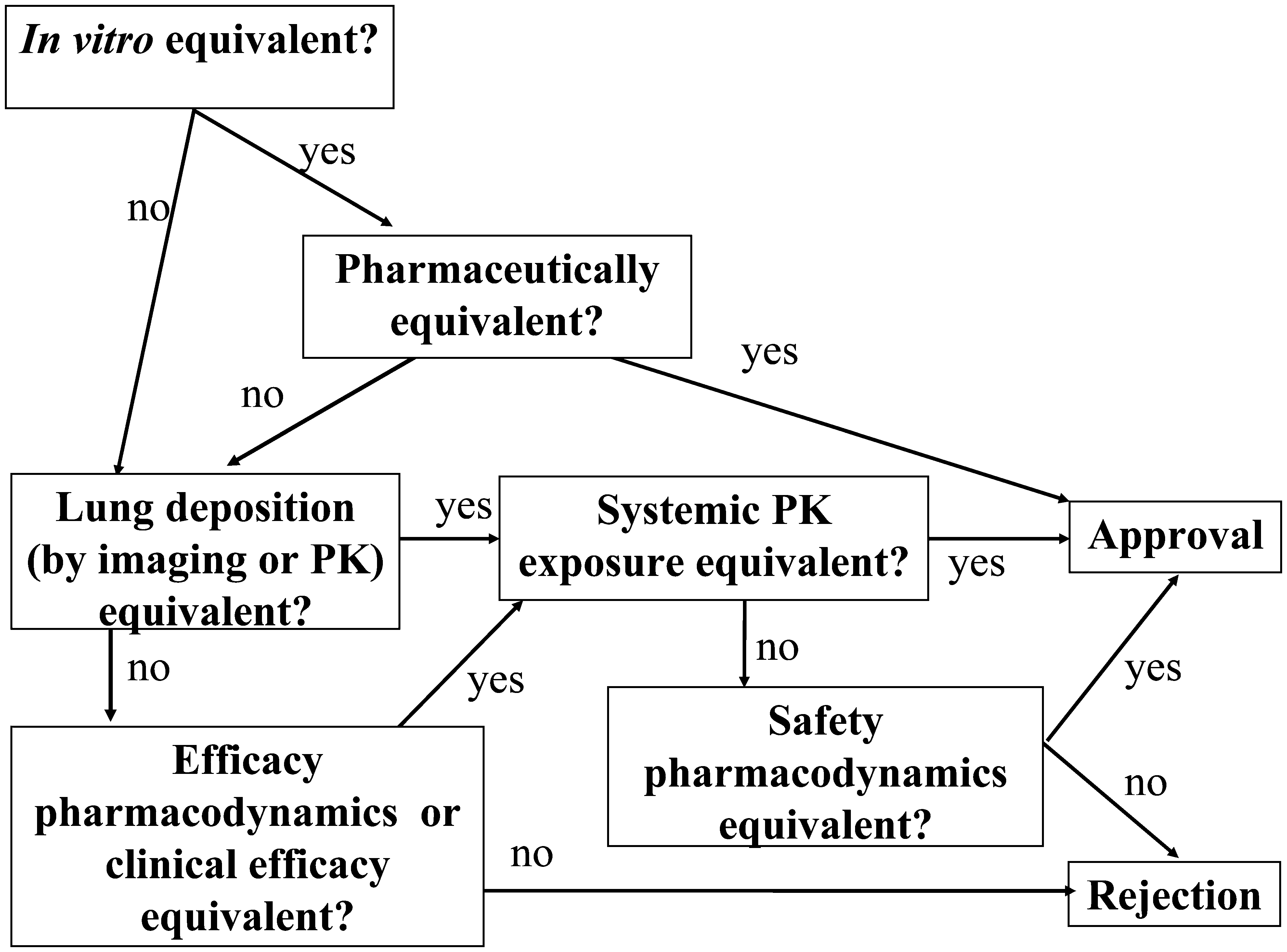
The stepwise approach for demonstrating therapeutic equivalence of OIPs for adults, as outlined in the EMA 2009 guideline. For children, the step “Lung deposition (by imaging or PK)” is not applicable.
In the stepwise European approach, comparative in vitro data from a number of tests (e.g., such as from a multistage cascade impactor or multistage liquid impinger) may be sufficient evidence of equivalence, but only if the OIPs being compared meet all of the criteria in the European guideline (see later in this report for a complete list).
If comparative in vitro data fail to show equivalence,(3) PK studies comparing systemic exposure (safety) and lung deposition (efficacy) can be employed to demonstrate equivalence of OIPs. In other words, in vitro results failing to show equivalence can be superseded by PK studies demonstrating equivalent exposure and lung deposition in vivo. If the comparative lung deposition and PK parameters meet the acceptance criteria, then the PK results are acceptable to support drug approval despite the in vitro differences between the T and R OIPs.
If in vitro and PK testing do not indicate equivalence, then PD testing is required. At the Workshop, some participants pointed out that relative to PK and in vitro data, PD data are more variable and potentially less sensitive to differences between products. They questioned why a sponsor would be asked to perform in vitro and PK studies at all if the lack of equivalence in in vitro and PK data can be superseded by the less discriminating PD or clinical studies. In response, European regulators explained that:
• PD studies are acceptable only if they are sensitive (i.e., can distinguish between two different doses—see later in this report for further discussion). • If in vitro and PK data do not demonstrate equivalence, the regulatory decision for OIPs may be based on PD/clinical studies for both pulmonary efficacy and systemic side effects, because differences in in vitro and PK characteristics might not impact clinical safety and efficacy. Thus, equivalence of in vitro and/or PK studies offer the opportunity to waive PD/clinical studies, but the lack of formal in vitro and PK equivalence calls for PD/clinical studies. • In vitro studies are the starting point for demonstrating equivalence in the EU. If in vitro equivalence criteria are not met, PK studies would be performed. The reason for going through these steps in this order (and not initiating PD studies right away) is that in vitro and PK results might show substantial product differences such that in vivo studies would be unethical or unlikely to succeed. Significant differences in in vitro and PK data may strongly argue for reformulation or redesign of the Test product. Conversely, continuation of clinical development might be justified and reasonable if in vitro and PK studies do not indicate substantial product differences. Further, PK study outcomes provide guidance on the nature of PD and/or clinical study requirements (i.e., determine the need for additional safety or efficacy PD studies).
In contrast to the EU where a stepwise approach is acceptable, the U.S. FDA and Health Canada in practice require all aspects of comparative testing, including in vitro, PK and PD (even though FDA has no formalized BE guidance for OIPs yet). For innovators working on product line extensions but using exactly the same device, source of APIs and excipients as the original OIP, the requirements might be modified in some respects. Of note, neither Health Canada nor the FDA currently allows use of PK data as a surrogate for PD efficacy; comparative PD efficacy studies are required in all cases. The rationale for the more conservative North American approach to pulmonary PK data appears to stem from concerns about the ability to quantify APIs in the systemic circulation resulting exclusively from pulmonary absorption(6) and concerns regarding the insensitivity of PK studies to differences in regional drug deposition that could affect efficacy.
Health Canada issued a guidance for second entry short-acting beta agonists (SABA) in 1999,(7) which does not accept “blood-level” studies “unless it can be shown that the analyte measured in the blood indicates what went through the lungs and its effect.” The Health Canada 2007 draft guidance for subsequent market entry of inhaled corticosteroids (ICS) for asthma(8) recommends determining the systemic exposure via PK studies; it also provides for systemic exposure to be determined in a pharmacodynamic (PD) study by assessment of the effect on the hypothalamic–pituitary–adrenal axis (HPA) if the plasma levels are too low to enable reliable analytical measurement. No specific in vitro acceptance criteria for bioequivalence purposes are included in the 2007 draft guidance, although that guidance requires “complete chemistry, manufacturing, and quality data” as well as “appropriate comparative data versus the Canadian reference product.” Pharmaceutical quality requirements were published in a joint Canadian–European guideline;(9) further information is available from an earlier Canadian guideline about preparing submissions in the Common Technical Document format.(10)
The FDA Office of Generic Drugs (OGD) has previously issued interim draft guidances for documentation of BE of pressurized metered dose inhalers (pMDIs)(11,12) and locally acting aqueous nasal aerosols and sprays.(13) In addition, FDA/OGD has provided insight into their expectations for demonstrating bioequivalence of OIPs through public meetings and publications.(1,2,14–17) Although there is currently no formal FDA guidance in effect on demonstrating BE for OIPs, based on the available information,(15,18) a “Weight-of-Evidence” approach is used by FDA. This approach incorporates qualitative (Q1) and quantitative (Q2) formulation sameness, device similarity (e.g., from the patient-use perspective), in vitro equivalence of the product performance, PK assessment of systemic exposure (safety), and PD assessment of local delivery (efficacy).
The differences in regulatory requirements between the EU, Canada, and the United States are summarized in Table 1.
Considerations for “In Vitro Only” Equivalence
The first step in the European guideline describes the possibility of in vitro equivalence testing as the sole surrogate for establishing therapeutic equivalence of OIPs. The relative ease of operation, the high power to detect differences between products, and the relatively low analytical method variability make in vitro testing an attractive option for establishing equivalence of OIPs compared to the other approaches (e.g., PD, PK, and imaging studies). Generic OIP approval based only on the in vitro data is in principle allowed in Europe, but up to now, only a very limited number of such approvals have been granted. During the Workshop it became evident that solution pMDIs and nebulizer solutions may have the highest probability of success following this pathway. By contrast, the T and R DPIs rarely interface with the patient identically; for example, due to the mouthpiece shape or dispersion mechanism. Before a company considers an “in vitro-only” submission in the EU, it should ensure that certain prerequisites are met, as summarized in Table 2, which also lists the hurdles that must be overcome in order to show that “pharmaceutical criteria for equivalence” are satisfied. A product must satisfy all of these in vitro pharmaceutical criteria for equivalence; otherwise, in vitro studies alone will not be considered sufficient for substantiating equivalence in Europe.
The comparative in vitro studies must be performed according to a preestablished protocol, including proposed batch selection, cascade-impactor-stage pooling, and in vitro acceptance limits. As these are frequently points of discussion during marketing authorization procedures in the EU, it is recommended that the protocol be preapproved by the appropriate regulatory agency.
Of all the in vitro requirements, aerodynamic particle size distribution (APSD) comparisons attracted the most attention at the Workshop. The remainder of this section focuses therefore on the APSD test.
APSD comparisons
APSD acceptance criteria
Comparisons between two OIPs are commonly made using inertial impaction methods, specifically either the Andersen cascade impactor (ACI) or the Next Generation pharmaceutical Impactor (NGI). The EU guideline requires the comparison to be performed per impactor stage or justified groupings of stages. The proposed acceptance criteria are: the 90% confidence interval of the T/R ratio must fall within 0.85–1.18 (where 1.18 is the inverse of 0.85) for all stages and/or groupings, although the applicant can propose and justify alternative limits.
The limits have been intentionally chosen to be restrictive, because of the limited availability of studies demonstrating a correlation between APSD data and in vivo performance. These restrictive equivalence criteria may be challenging to meet when comparing individual stages between T and R, due to the high variability of APSD measurements, especially in the case of very low API amounts on stages. A simulation study presented during the Workshop(19) demonstrated that a hypothetical product with between-batch and within-batch variability typical of approved OIPs could not be shown equivalent to itself when two sets of batches were compared following the methodology of the EMA guidance. This suggests that the acceptance criteria mentioned in the EMA guideline may be too stringent, at least for some stages/groupings. Taking the variability of the R as a starting point for establishing more realistic acceptance limits (and they may be different for different stages) was proposed as a better alternative. It could be instructional to see results of a study using real data for OIPs currently available on the market (to the best of the authors' knowledge, such a study has not been conducted yet).
Another question related to the EMA APSD criteria is whether the acceptance limits should be applied to the T/R ratio of API depositions, or to the absolute differences in API deposited on each stage. Furthermore, it was argued that the best approach may be a metric that in a scientifically valid way combines the stagewise differences into a single parameter that measures goodness of fit between the T and R particle size distributions. An analysis of one such approach(20) had been conducted within PQRI, with the conclusion that the chi-squared ratio metric is not sufficiently discriminating;(21–23) however, no alternative recommendations were made by that group due to the agreed constrictions on the scope and time for that project.
Batch selection
The unsuccessful result from the above-mentioned simulation study is not solely due to the difficulty of showing equivalence for stages with low load and therefore high variability. Another major reason is the between-batch variability, either due to batch-to-batch inherent differences or due to changes over time during storage. Because typically only three batches of T and R are studied in the in vitro equivalence experiments, there is a risk that all three batches of R come from one end of the normal range. The strategy for batch selection should be developed in consultation with the appropriate regulatory agency. This batch-selection problem is a consideration for all equivalence testing, not just APSD testing.
Stage groupings
If impactor stages are grouped for the data comparison, the EU guideline recommends at least four groups of stages, and expects the grouping to be justified by the expected deposition sites in the lung. In practice, grouping is typically done for adjacent stages and frequently justified by linking the selected impactor groupings to sections of the airways (e.g., mouth/throat area, upper and lower parts of the lung). However, impactors differ in many aspects from the airways(24,25) and studies investigating in vitro–in vivo correlations are largely absent or contradictory and nonconclusive. The utility of these justifications is therefore debatable. Another aspect of discussion at the Workshop was the fact that groupings may hide information that may be relevant for the equivalence assessment. In practice, stages with very low amounts of drug substance are frequently grouped with other stages, because these low amounts are claimed to be irrelevant for the clinical outcome. Although this assumption seems to be reasonable, it is difficult to prove that a small amount of drug deposited on a stage is irrelevant. Another reason for grouping stages with low API load is that the relative variability typically is inversely proportional to the load, making the likelihood to demonstrate equivalence very low even when the T and R truly are equivalent.
During the Workshop's breakout session on in vitro testing, participants discussed whether some form of abbreviated impactor measurement (AIM)(26,27) could be viewed as representing deposition in the throat, central, and peripheral areas of the lung—at least in one population group, for example, an average young, healthy Caucasian male. It was pointed out however, that although AIM in combination with efficient data analysis (EDA)(28–31) may be more discriminating in detecting small differences in APSD (and thus useful for quality control purposes, to ensure batch-to-batch consistency of APSD), the in vivo relevance of such differences, as well as in vivo relevance of the selected AIM cut points, remain unproven and will need to be demonstrated for any given product.(32)
Alternative particle sizing techniques
The use of cascade impaction as a quality control tool has been well established since the late 1980s.(33) However, an ACI, described in the European Pharmacopeia (EP),(34) had originally been designed to collect airborne microorganisms(35) and was only later coopted for characterization of pharmaceutical aerosols. Thus it was not developed with the end goal to maximize usability in the pharmaceutical laboratory, nor to possess in vivo relevance.(33) Other impactors were developed specifically for particle sizing of pharmaceutical aerosols, including the next generation pharmaceutical impactor (NGI) and Marple-Miller impactor (MMI). Although both are specified in the USP,(36) only the NGI is recommended by the EP.(34) The NGI possesses a comparable number of stages to the ACI, whereas the MMI has fewer stages and so may not be as discriminatory in the very fine particle size range (although this conclusion is now partially challenged by the findings supporting the AIM/EDA concepts). Other acceptable EP sizing devices include Apparatus A (the two-stage glass impinger), Apparatus B (the metal impinger), and Apparatus C (the multistage liquid impinger), each of which has fewer “stages” than an ACI or NGI. In theory, more stages should provide more information about APSD. However, because the total amount of API is finite, fractionating the dose into more than four to five size ranges may degrade the quality of information.
Other sizing techniques, not involving impactors or impingers(37) exist but have not been used for equivalence testing, although they might be useful for product development and characterization, especially for solution-based OIPs.
In vitro–in vivo correlations (IVIVCs) for APSD
Flows and throats: in order to ensure consistent impactor performance in the size ranges of interest, the cascade impactor is operated over a limited range of fixed flow rates. Unfortunately, the stage cutoffs for a situation when an impactor is used with a realistic inspiratory profile have not been established. This means that standard impactors cannot be operated in a way that is reflective of the patients' inspiratory profiles, which vary both in flow rate and time profile. This limitation may be of particular relevance for the comparison of DPIs, where differences in device airflow characteristics and dose release mechanisms may exist depending on the flow rate. In such instances, it may be more appropriate to use patient-relevant flow rates and flow acceleration to characterize inhaler performance. One way that this can be achieved is to use simulation equipment that mimics an average patient's inspiratory profile through the inhaler, but also interfaces to the cascade impactor (CI) in such a way as to permit sizing at constant flow rate appropriate to the impactor. This approach has been adopted in such instruments as the Electronic Lung(38) or the Pari simulator.(39) In addition, modification of the impactor induction port to more closely model in vivo oropharyngeal deposition has also been investigated; such approaches include use of oropharyngeal casts(40–42) or more idealized throats such as the Alberta Throat.(43) It is possible to combine both realistic mouth–throat models and realistic inhalation flow rates and then size fractionate the aerosol exiting the throat in a cascade impactor at a constant flow rate.(44) However, at present there is no standardized approach for such measurements, and many of these models may be too complex for either equivalence comparisons or routine QC testing.
Mathematical modeling: development of in vitro methodology to predict in vivo deposition remains of widespread interest.(45) Although CIs are an accepted QC tool to characterize APSD, they were never designed to simulate deposition in the human respiratory tract, and in fact, the collection efficiencies for various ACI stages differ dramatically from those for regional lung deposition.(24,25) APSD determination in a CI is predominantly based upon particle deposition due to inertial impaction whereas the situation in the human respiratory tract is much more complex and involves inertial, sedimentation and diffusion mechanisms. Once the aerosol is generated, the individual particles may also change in size or shape due to hygroscopic growth or evaporation during transit in the respiratory tract—but this is difficult to model or predict.
Several paths have been proposed to predict in vivo lung deposition from in vitro APSD data. An accurate determination of the in vitro APSD characteristics of the aerosol that reflects realistic in vivo conditions (taking into account flow rate, temperature, humidity, and airway geometry) is key. One can then, in principle, apply a mathematical model to predict in vivo deposition. At a minimum, the prediction should include at least three regions: the extrathoracic, bronchial, and peripheral or alveolar regions. Another scenario would be to use a “realistic and standardized” throat and lung (which do not exist at present), to quantify the deposition in the various regions of the respiratory tract. Combinations of these strategies can also be envisioned. For example, as described earlier, a realistic inhalation flow profile and throat model could be used to quantify extrathoracic deposition and then a CI be used to measure the APSD. The amount of drug deposited in the CI gives a reasonable estimate of the total amount of API deposited in the lung but if prediction of deposition in various regions of the lung (e.g., the bronchia and alveoli) were desired, then a mathematical deposition model would be required in addition to CI data. Part of the mathematical consideration would have to be the different flow rates and other parameters affecting in vivo deposition. A further approach would be to correlate APSD data with in vivo PK outcomes (Cmax, AUC, etc.) as a reflection of the total and regional lung deposition. Such studies have met with limited success.
Correlating APSD and imaging: total and regional deposition can also be measured using OIPs labeled with radiotracers and imaging. However, these studies are limited in part by the resolution of the scanning equipment and the need to use a surrogate product (to create the radio-labeled product). In addition, small ventilatory changes during inhalation of the labeled drug aerosol, as well as changes in airway geometry due to disease, mean that the practical clinical relevance of such studies is limited.
Individual patient IVIVC?: Matching in vitro APSD profiles does not ensure equivalent in vivo deposition, dissolution, and clinical effects. Even if an IVIV relationship were established for APSD, such a general correlation might not be valid for a given patient's airways' anatomy, physiology, and disease state. Predictions of in vivo deposition will only apply to an average healthy adult or child. The patient–device interaction can additionally cause significant variability in performance of OIPs and consequently “IVIVC” between individual patients.
Ultimately, what is important is the safety and efficacy of the deposited dose, and so the deposition information would need to be linked to information on the site of action and/or the location and distribution of the receptors of interest. Even if the in vitro and in vivo deposition profiles are shown to be the same between T and R, differences in the physicochemical properties between T and R drugs could result in changes in the dissolution profile in the lung, which has the potential to change its safety and/or efficacy. Thus, acceptance criteria describing allowable differences in APSD profiles may need to depend on the specific drug, disease, and disease state being treated.
Considerations for PK Studies in Establishing BE of Locally Acting OIPs
The objectives of PK studies for demonstrating BE of locally acting OIPs are twofold: (1) comparison of total systemic bioavailability (as an indicator of safety, to ensure that T and R do not differ in systemic side effects), and (2) comparison of the pulmonary bioavailability (as an indicator of efficacy). In the European stepwise regulatory approach, PK studies that demonstrate equivalent pulmonary deposition combined with adequate safety data may be considered sufficient to establish therapeutic equivalence. Note that the term “bioequivalence” is often used in EMA guidelines when discussing PK, and will therefore be used in this section.
The EMA requires double-blind, crossover PK studies with clinically relevant doses and strengths in the intended adult patient population. Studies in children are to be limited to the assessment of the systemic safety, whereas efficacy (pulmonary equivalence) is to be addressed by PD or clinical studies (see further discussion of this issue later in this report). Within PK studies, the EMA recommends standard BE methods and metrics (Cmax, AUC). In addition, the time at which Cmax is observed (tmax) is included as an additional outcome parameter for OIPs, which is not generally mandatory for solid oral dosage forms in Europe,(3,46) except in those cases where the rate of absorption has significant clinical importance.
Studies as a marker of systemic exposure (safety)
There is a general consensus that PK studies assessing total systemic bioavailability(3,8,16,47) are a valid surrogate for PD safety effects. This is in keeping with standard PK principles used to assess BE of T and R (e.g., for oral tablets). If T exhibits greater systemic bioavailability than R (i.e., outside the recommended limits), additional safety PD studies are usually required to ascertain if the additional exposure is associated with any changes in the drug's systemic safety profile. Conversely, if a systemic PK study demonstrates that exposure with T does not exceed that of R, a waiver of safety PD evaluation could be discussed with the appropriate agency.
North American attendees questioned the rationale for allowing PD safety studies within the stepwise BE approach (per the EMA guideline) for situations where PK-based systemic BE failed (i.e., T exposure is higher than R exposure). The European regulators explained, however, that PD studies may demonstrate that the observed PK differences lack clinical relevance. In the case of higher systemic exposure levels achieved with T, the EMA would therefore accept properly designed and sensitive safety PD/clinical studies (e.g., 24-h plasma cortisol suppression), and may conclude equivalence if the observed PK differences between T and R do not translate into clinically meaningful differences in safety characteristics. It was suggested that widening the PK acceptance limits based on well-established PK/PD relationships could be an additional way to justify that the PK differences lack clinical relevance.
PK studies as a marker of pulmonary deposition (efficacy)
The European guideline requires demonstration of equivalent pulmonary deposition, as a surrogate for efficacy. This could be done either by lung imaging (gamma) scintigraphy of the radiolabeled OIP or by PK efficacy studies (Fig. 1). Note that lung imaging cannot replace PK safety (or PD safety, if needed) studies. Furthermore, neither PK efficacy nor lung imaging studies are allowed in children, so PD or clinical studies are needed to demonstrate equivalent efficacy in children, per the EMA guideline.
The PK studies for OIPs are quite different from the traditional PK bioequivalence paradigm. Although it is accepted in the scientific community that PK studies of OIPs are the method of choice to probe for differences in the systemic exposure, the ability of PK studies to identify differences in the pulmonary deposition characteristics (defined as the extent and pattern of pulmonary deposition of an inhaled active substance) is still being debated. A key question is whether PK studies are suitable for comparing the efficacy of OIPs.(6,48) This is because plasma drug levels may not necessarily represent the drug concentration at the target site within the pulmonary system and do not determine the effect at the end organ of interest, that is, the pulmonary effect. Rather, plasma concentrations of an OIP drug reflect end-organ deposition measured in a compartment downstream of the target organ.
In PK studies involving drugs that do not have appreciable oral bioavailability (BA) or that are performed under conditions that do not allow oral absorption (e.g., due to a charcoal block) of the swallowed fraction of an orally bioavailable drug, the lung is the only compartment from which the drug is absorbed into the systemic circulation. Under such conditions, systemic plasma concentration time profiles may provide information on the absorption processes in the lung (deposited fraction of the dose, particle size distribution pattern, pulmonary retention time, dissolution processes, and central-to-peripheral deposition pattern).
For OIPs with appreciable oral bioavailability, PK efficacy studies need to be designed in such a way that they allow assessment of the pulmonary deposition. Two methods were discussed at the Workshop: the charcoal block method, which prevents gastrointestinal (GI) absorption of the swallowed fraction of drug; and the “early” bioavailability method, which provides information about lung deposition if absorption from the lung is faster than from the GI tract.
The EMA allows pulmonary PK data to serve as a surrogate for efficacy,(3) that is, demonstration of BE in a pulmonary PK study may preclude the need for subsequent PD efficacy studies. If pulmonary deposition is lower for T than for R, evaluation of comparative efficacy by PD or clinical methods is required. If pulmonary deposition is higher for T than for R, additional clinical safety data may be required, along with further comparative consideration of in vitro characteristics (such as comparative APSD, especially FPD data) in order to evaluate whether the observed PK differences can feasibly be associated with clinically relevant differences of regional lung deposition pattern.
Support for the European regulatory approach stems principally from the few published studies that simultaneously evaluated pulmonary PK profiles and PD efficacy of different formulations/doses of OIPs.(49–53) These studies—all focusing on β-2 agonists—demonstrated a relationship between increasing lung dose and greater pulmonary effects. Literature reviews incorporating cross-study comparisons have also concluded that a similar relationship exists.(54,55) It is possible that a particular drug is at the plateau of a dose–response curve, so a greater or lower drug deposition in the lung may not necessarily mean changed clinical response; but this question will have to be resolved through PD or clinical studies, which are required by the EMA if equivalent pulmonary deposition is not demonstrated.
Several other publications suggested that for ICS as well, PK studies can reflect the local deposition of OIPs at the site of action. For example, PK studies of a beclomethasone dipropionate formulation showed higher pulmonary deposition in an HFA compared to the CFC product,(56) in agreement with the protective effects found in PD studies. Likewise, similar protection against methacholine-induced airway hyperresponsiveness (methacholine PC20) in asthmatic patients provided by HFA and CFC formulations of budesonide was mirrored by their equivalent PK profiles in healthy adult subjects.(57)
Two other studies showed that PK studies are more sensitive than PD or clinical studies in identifying product differences. Daley-Yates reported that neither in vitro data nor a 12-week clinical study in asthma patients were able to demonstrate differences between two DPI products combining fluticasone propionate (FP) and salmeterol (SAL), whereas a PK study displayed differences.(58) Similarly, the authors of a recent study comparing two DPI FP/SAL combination products concluded that their results “… underscore the difficulty in relying on pulmonary function to compare inhalation products intended to treat asthmatic patients,”(59) as PK studies showed the higher discriminatory power.
Charcoal block
For the charcoal block approach, the requirement to validate the effectiveness of the employed charcoal block for a given API(s) was emphasized. Validation can be accomplished either by referencing published evidence, or by the applicant's own (in vitro or in vivo) validation studies. At the Workshop, concerns were expressed that oropharyngeal mucosal absorption could confound PK-based pulmonary delivery assessments. This concern, however, was countered as being largely hypothetical considering the small oropharyngeal mucosal surface area (compared to the bronchial and GI tract surface areas) and the generally short drug residence time, given that oropharyngeal deposits are likely to be rapidly swallowed.(60) Furthermore, mouth rinsing, which is recommended for all ICS-containing OIPs, would effectively minimize the fraction of the dose that feasibly can be absorbed via this route.
Another concern with charcoal block methods was the possibility that the ingested charcoal may not only prevent the absorption of the swallowed fraction of dose, but may lead, for certain drugs, to a more complex interference with their overall disposition, for example, by disrupting enterohepatic recycling of drug amounts undergoing biliary or intestinal secretion. It was argued, however, that in general, this mechanism should not compromise the comparisons of pulmonary deposition, because it would likely affect the T and R formulations in a similar way.
Early bioavailability
In principle, the measure of early exposure by assessing partial AUC values (e.g., 0–30 min postdose) could be an alternative approach for estimating pulmonary drug exposure, and it has already been employed by the FDA in the approval of generic albuterol MDIs. However, it was stated at the Workshop that a simple and straightforward general translation of this approach to other compounds is not applicable, as different absorption properties (in particular, the rate of absorption characteristics) will require specific time windows to be established to accurately capture the fraction of the dose absorbed from the lung. Hence, it was generally agreed that the early-BA approach will need to be validated for each API and formulation in question, and will depend on their particular PK characteristics. It was further mentioned that for solid oral dosage forms, it had already been shown that early partial AUCs display higher variability as opposed to Cmax values.(61) This is a separate issue deserving careful exploration before any recommendations in this respect can be made.
Using PK parameters to describe pulmonary fate of OIPs
In general, data from PK efficacy studies can provide the following information about a drug's pulmonary fate: (1) total pulmonary dose and absorption, (2) deposition pattern in the lung (e.g., central vs. regional), and (3) absorption rate and residence time in the lung (the latter parameter is not explicitly discussed in the EMA guideline).
There is no controversy regarding total pulmonary deposition, as numerous studies(56,62) have demonstrated that PK is sensitive to differences in the deposited dose, and thus, PK is able to differentiate between two products that deliver different drug amounts to the lung.
PK data can reflect not only the total amount but also the rate of pulmonary absorption of drugs that are not orally bioavailable or where oral BA is precluded, for example, by a charcoal block. The rate with which the inhaled drug is absorbed from the lung into the systemic circulation may contribute to systemic side effects, but it may also provide information about APSD characteristics and the time of residence of deposited drug particles in the lung. These properties are known to affect the pulmonary efficacy and the degree of pulmonary targeting. The EMA guideline suggests that in accordance with the standard accepted methods of BE assessment, the maximum concentration (Cmax), the area under the curve (AUC), and the time to Cmax (tmax) be compared.
Nevertheless, participants were divided as to whether PK studies are generally suitable to assess the fate of the drug at the pulmonary site of action. Some participants expressed the concern that PK studies are unable to fully assess the pulmonary fate of the drug, stemming primarily from the view that unlike conventional PK measurements, where plasma concentrations are considered the key determinant of product-related target organ exposure characteristics, plasma concentrations for OIPs correspond to a downstream compartment relative to the target organ (the lung). Thus plasma concentrations were considered to be the result/product of pulmonary fate (assuming oral BA is negligible or blocked by charcoal), rather than the determining factor for lung concentrations and therefore lung effects. Other participants believed that standard PK parameters such as AUC and Cmax are the result of the pulmonary deposition properties of OIPs and that PK can be used to adequately assess potential and clinically meaningful differences in lung deposition between two products.
EMA shares the view that PK can provide information on “the extent and pattern of pulmonary deposition of an inhaled active substance”(3) and the EMA guideline proposes the use of PK studies to probe for the ‘pulmonary deposition’ of OIPs in adults, if GI absorption is blocked or negligible. At the same time, the EMA guideline states that “limitations with PK studies include their inability to differentiate the distribution of drug within the different zones of the lung.” Overall, further research is required to confirm the potential for, and limits of pulmonary PK studies to detect regional lung drug deposition patterns and to ascertain whether formulations with similar in vitro characteristics and equivalent PK can feasibly exhibit different pulmonary effects. Additional data examining the relationship between pulmonary PK and pulmonary effects may aid the development of an internationally harmonized consensus position on this issue.
Within this context, the ability not only of PK but also of PD or clinical studies to distinguish between formulations that deliver the same dose to different regions of the lung was also questioned. As a result, it was one of the conclusions of this meeting that the suitability of PK and clinical studies in detecting differences in pulmonary deposition pattern represents a knowledge gap and that more studies are necessary to compare the robustness and sensitivity of PK results for formulations that only differ in the regional deposition pattern. Such studies could compare such products in PK, as well as in PD or clinical studies, and evaluate which method shows higher sensitivity and robustness in identifying formulation differences, along with quantitative thresholds indicating clinical relevance. A careful evaluation of such results would hopefully inform pharmaceutical and clinical development concepts and aid regulatory decision making.
Points to consider about tmax
Probing for differences in tmax, although at first sight seemingly relevant for assessing the rate of absorption, appears problematic, because of the noncontinuous nature of this parameter (due to measuring only at the preset time points, e.g., 5 min, 10 min, 30 min, etc., even though a true tmax may fall between those times). If a nonparametric statistical approach for tmax is applied, as appropriate and mandatory, even for T and R showing the same deposited dose and absorption kinetics, a PK study using a sample size sufficient to demonstrate BE for AUC and Cmax, would have a risk to fail merely based on additional BE requirements for tmax. Therefore, some have argued that this parameter should not be used within the BE assessment.(63)
Indeed, it is currently regulatory practice not to employ a strict statistical BE evaluation of tmax outcomes, but merely to compare medians for tmax, whereby comparable medians (e.g., a match within adjacent, rather than identical, time points on the blood sampling schedule) are considered similar. However, the rationale for this additional BE requirement for OIP products remains largely unclear, considering the inherent methodological obstacles with the noncontinuous (i.e., discrete) nature of this parameter, rendering its outcome entirely dependent on the PK-sampling schedule employed. The need for tmax comparisons is especially doubtful because Cmax is a more sensitive and discriminatory performance metric for the rate of absorption.(63)
What Cmax measurements can tell us
Potential differences in the pulmonary residence time (e.g., due to differences in the dissolution rate of deposited particles) will be identified in PK studies through parameters sensitive to the absorption rate, such as the maximum observed concentration (Cmax), which depends not only on the pulmonary delivered dose but also on the absorption rate from the lung.(64–66)
Although use of Cmax is accepted for assessing differences in absorption rate of drugs with moderately fast, intermediate, or slow absorption rates, specific challenges were discussed related to ensuring reliable estimates for very rapidly absorbed drugs, such as long-acting beta agonists (LABAs), which are often used within multiple API products. These challenges include the proper selection and number of time points early after dose administration and the relation between the sampling frequency and associated variability. With regard to the LABA components of multiple API products, the very rapid rate of absorption of formoterol and salmeterol was emphasized, which often results in the situation that Cmax is observed already in the first blood sample (frequently taken 5 min postdose because of the perceived need for multiple inhalations for the proper quantification of systemic LABA exposures). The potential implications of the regulatory acceptability of the “first-sample Cmax data” in lung-deposition BE PK studies were discussed, but no consensus was identified. Attending regulators emphasized that efforts should be undertaken by the applicants to manage an earlier initial blood sample (e.g., at 2–3 min) to more accurately capture maximum exposure levels (i.e., Cmax). However, the need for multiple inhalations to achieve quantifiable systemic exposure over a sufficient amount of time may still make it difficult to accurately capture Cmax despite those efforts.
Information on the regional (central vs. more peripheral) lung deposition of OIPs can also be obtained from PK studies (specifically, Cmax values), with appropriate modeling. For low pulmonary permeability and/or slowly dissolving drugs, combined with more central lung deposition characteristics, a more efficient ciliary removal of the centrally deposited fraction of dose can be expected.(67) In turn, products delivering highly permeable and fast dissolving compounds and/or achieve more peripheral lung deposition to smaller airways are expected to result in higher Cmax values(68,69) due to faster absorption.
Study populations
Patients versus healthy subjects
The EMA OIP guideline suggests conducting PK studies “in the intended patient population.” This requirement was subject to debate at the Workshop because BE studies should be performed in a population displaying the highest probability for discrimination between T and R. PK studies in asthmatic patients display higher variability than those in healthy subjects, because of disease-related changes/variations in airway caliber and bronchial hyperresponsiveness. In line with these considerations, PK studies in asthmatic patients have been shown to display higher variability than those in healthy subjects.(70) The actual degree of airway obstruction in asthmatic patients was shown to determine the total extent of pulmonary deposition as well as regional deposition pattern (i.e., predominant deposition in central airways at conditions of pronounced bronchial obstruction, indicated by low FEV1). These observations are supported by another study in asthmatic subjects, which reported a significant positive correlation between FEV1 and pulmonary deposition of beclomethasone and mometasone furoate.(70) The overall tendency toward a more central airway deposition in asthmatic patients, therefore, generally has a potential to compromise the proper characterization of product-related differences in their regional airway deposition characteristics (i.e., central vs. peripheral deposition).
Furthermore, studies in patients are often associated with ethical constraints (i.e., washout of established treatments) and numerous confounding factors. Overall, the available data suggest that PK studies in healthy subjects are likely to be at least as, or more, sensitive than patient studies to detect performance differences between different products. The use of healthy subjects avoids the potentially confounding influence of variable airways obstruction across study periods in asthmatic subjects. In summary, it was considered reasonable to preferentially conduct OIP PK studies in healthy subjects. Indeed, there is already regulatory precedent in Europe to allow studies in healthy subjects (see EMA's home webpage, information about past approved products, public assessment reports).
PK studies in children
Physiological and anatomical differences between airways of children and adults affect the handling and performance of pMDIs and DPIs. EMA therefore generally requires in vivo studies in children for the BE assessment. As challenges associated with the time coordination between pMDI drug release and the child's inhalation are generally solved in children by using pMDIs in conjunction with spacers, pMDI approval in children is linked to coapproval of specific spacer device(s) that need to be included in in vivo studies in specific age groups (see below).
In contrast to pMDIs, DPIs have the advantage of automatically coordinated inhalation and drug release. However, performance of DPIs depend on device resistance and inspiratory flow rate characteristics, which again can result in product performance differences between pediatric and adult populations. For this reason, EMA requires a balanced enrollment and representation of children across the relevant age categories intended for use, for example, <2 years, 2–5 years, and 6–12 years.
The EMA specific recommendations for PK BE studies in children differ, however, from those in adults.(3) PK studies are only recommended to evaluate the systemic safety in children, but not the pulmonary deposition, as is the case for adults. The EMA guideline states that pediatric PK efficacy studies are inappropriate because they might increase the experimental burden on the child while only indirectly implying efficacy. Some Workshop participants speculated that the EMA might also be concerned about methodological difficulties of charcoal studies with serial PK assessments in pediatric populations.
This conservative position regarding PK studies in children, based on ethical concerns regarding frequent and numerous blood draws in children, is understandable. These concerns could be overcome, however, for example, by less frequent sampling around Cmax, and/or by using population PK approaches, which use more children but fewer blood draws per child.(71) The employment of noninvasive sampling approaches of other biological matrices (e.g., saliva) could be another solution. These alternatives were unfortunately not discussed in the 2009 EMA guideline.
The recommendation to employ PK BE approaches in children only for the assessment of safety but not efficacy, seems to contradict EMA's position for other dosage forms, for which full PK studies in pediatric populations are frequently used, especially because PK studies can mitigate the need for long-term clinical studies.
Despite these reservations, participants discussed the necessity of pediatric PK studies. Test OIPs might differ in their device design and handling, as well as in the airflow dependence of their deposition characteristics. Considering that T and R might differ in their in vitro profiles, for example, APSDs at the clinically relevant flow rates, the incorporation of PK studies could become necessary for DPIs with flow rate-dependent deposition characteristics, in pediatric age groups that are unlikely to achieve the range of adult inspiratory flow rates.
PK studies with multiple API products
Bioequivalence studies for products with two or more APIs are a challenge, because of the necessity of multiple comparisons and the difficulty of assessing the effects of individual active components in a product. During the Workshop, the participants discussed whether for multiple API products with comparable in vitro APSDs of both APIs, both components need to be compared via PK studies. Achieving formal PK equivalence for multiple API products under a variety of different conditions (with and without charcoal, with and without spacing devices for pMDIs, and at different flow rates for DPIs) is a significant challenge. It was noted that for products containing two active components, there would be in total at least eight PK parameters to meet BE acceptance criteria even without considering tmax requirements (i.e., AUC and Cmax for each of the two APIs, with and without charcoal each). The chance of meeting the 0.8–1.25 acceptance criteria for each of the eight comparisons might be low. The Workshop participants highlighted these resource and statistical challenges, and encouraged regulators to consider them.
Some participants suggested that the systemic exposure of one active component could be reasonably interpreted as reflective/indicative for the pulmonary delivery of the second active component, as long as the in vitro characterization (APSD and other in vitro tests) are indicative of a reasonable codeposition of both active components in the airways (e.g., comparable fine particle fractions and similar stage-by-stage APSD). For instance, FP, which is not absorbed to a significant extent from the GI tract, could serve as primary and adequate/sufficient indicator of the lung deposition characteristics of an FP/SAL OIP, without the need for a separate PK comparison of the LABA component, whereas the LABA equivalence could be assessed through PD studies. This approach would further offer the opportunity to waive the need for charcoal block PK studies for products containing fluticasone as one of the components. Overall, no consensus was reached and further studies in this area appear necessary.
If PK studies fail to demonstrate equivalence, it might be difficult in the next step (PD/clinical studies) to identify suitable study designs and sufficiently discriminating clinical endpoints that would allow for a reliable assessment of the pharmacological effects of the steroid component of a multiple API product, because of the usually shallow dose–response characteristics and the slow onset of action of ICS. Some participants initiated a discussion as to whether PD studies might be necessary for both components if PK-based BE goals were not met or whether a widening of the PK range (e.g., for Cmax, that shows often higher variability(72) for the steroid compound) would be adequate. It was proposed that a combination of widening the PK acceptance range and assessing the PD of the component with the steeper dose–response and faster onset of action, might be sufficient.
Cmax, tmax, and AUC acceptance limits (Goalposts) for PK BE studies
The conventional BE limits for PK metrics (0.8–1.25) have been adopted in the European guideline for OIP products from the historical limits long applied to define BE requirements of other (e.g., solid oral) dosage forms designed for systemic action. [As discussed below, the EU guideline acknowledges that narrower or wider limits might be needed for narrow-therapeutic-index (NTI) or highly variable drugs, respectively.(3)] However, the in vitro release and through-life specifications for approved OIPs typically allow fine particle dose (FPD) variance of±20% to±45% of the mean, given methodological complexities associated with the quantification of FPD. Such in vitro variance is highly likely to preclude the attainment of an in vivo point estimate (let alone 90% confidence intervals) within the 80–125% range for T when tested against certain batches of R versus other batches of R.(73) Batch-to-batch variability of R was therefore a topic of discussion at the Workshop.
It appears rational to consider methods for scaling BE limits for OIP PK studies (beyond those already existing in the EU regulatory paradigm) by using statistical approaches that may allow scaling based on the R variability. Predefined differences in PK estimates will not necessarily translate into similar differences in clinical or PD studies. The EMA defined the allowable range of relative potencies of 67–150% for dose–response PD studies. This would translate into a much broader (than 67–150%) acceptance range for PK parameters if accepted PK/PD correlations are applied.(74) In turn, T products that are slightly outside the PK BE goalposts, are likely to demonstrate PD-based equivalence by a well-powered PD study. Methods for scaling BE limits for PK studies should therefore be considered and incorporated within the future paradigm. (The current EMA approach allows scaling only for Cmax of highly variable drugs.) With appropriate scaling, it may be possible to waive clinical or PD studies in some instances in the future.
The Workshop participants pointed out that in case of PK studies, for which the T-to-R point estimate is close to unity (meaning that PK means are similar for T and R), but the confidence intervals are too wide (i.e., extending outside the goalposts), the PK study should be repeated with a larger sample size and proper training of the subjects' inhalation maneuvers. If the point estimates have been shown to be substantially different, however, it was suggested that it might be necessary to redesign the T formulation or device characteristics and repeat the PK study. Some attendees suggested, however, proceeding with PD studies instead, assuming that PD or clinical studies would not necessarily reflect the differences indicated by PK data. Other attendees argued that when PK showed differences, moving directly to PD/clinical studies might not be ethical. The extent of T-versus-R exposure differences in single-dose PK studies that would make a PD/clinical study unethical was not specifically discussed.
The European guideline indicates that acceptance limits for systemic safety assessment might be narrowed for NTI drugs. Conversely, the limits for Cmax (but not AUC) could be widened to 0.75–1.33 for products with demonstrated high PK variability, that is, the study design must be of replicate design and the intraindividual variability for the R must be more than 30% (CV% ANOVA), and where such widening would not have clinical relevance, which may be difficult to prove for an OIP.
The possibility of narrowing the acceptance limits for NTI drugs was discussed in detail at the Workshop. So far, only glucocorticoids, such as FP may have been perceived by some individual stakeholders to be potential candidates for NTIs. However, the shallow dose–response and associated clinical practice with these drugs (i.e., typically doubling or halving of doses in order to titrate to effect) clearly argue against the categorization of glucocorticoids, including FP, as NTIs. PK/PD simulations for glucocorticoids such as FP given at clinically relevant doses, indicated that AUC differences of −20 or +25% (T vs. R), will alter the average cortisol suppression by less than 4%.(75) Considering these relationships, there does not seem to be a rational scientific basis for tightening the acceptance limits of inhaled corticosteroids.
General Considerations for the Design of PD Equivalence Studies
Regulatory requirements concerning the design of PD equivalence studies currently differ among the EU, Canada, and the United States (see Table 3). The EMA requires that at least two dose levels of generic and reference OIPs are compared for all drug classes whether in comparative efficacy or safety studies.(3) Health Canada and FDA adopt a similar approach for efficacy and safety studies of SABAs,(7,15,16) although in the case of FDA this appears to be a de facto approach because the FDA Albuterol MDI guidance was withdrawn in 1996. For ICSs and LABAs, Health Canada advocates comparison of only a single dose level of T and R versus placebo in order to assess comparative efficacy.(8,76) The Canadian ICS draft guidance states that the proposed study design is based on the assumption that the primary endpoint is sputum eosinophils. In this scenario, one dose is sufficient; however, no specific dose selection statement has been made regarding a study where FEV1 is used as the primary endpoint. In this scenario, Health Canada usually requires two doses of ICS to be tested; furthermore, the Canadian SABA guidance also requires two doses of SABA (one and two puffs).
A further difference in regulatory approaches between regions concerns the doses to be compared; for SABAs, Health Canada recommends the use of the “usually prescribed dose and doses higher than normally prescribed”—in safety studies and in efficacy studies, if required,(7) whereas the EMA requires that the highest approved dose be evaluated in comparative PD safety studies.(3)
(Bio)Assay sensitivity
In attempting to rationalize the regional differences, it is useful to consider the fundamental requirements of a study intended to evaluate equivalence between two products. The principal and overriding requirement is that treatment effects measured in a study be sensitive to differences in delivered dose or potency, that is, the study must have assay sensitivity.(77) To assure that a study satisfies this basic requirement, it has long been appreciated that an equivalence study must include at least two doses of T and/or R OIPs(78,79) enabling a within-study assessment of dose–response. If there is no dose–response information, then the study may lack the sensitivity to differentiate formulations, and therefore no inferences regarding equivalence may be made.
Despite this fundamental tenet, the comparison of single dose levels of T and R alongside a placebo arm has been advocated in recent years as an adequate regulatory approach by Health Canada.(8,76) This approach provides assay sensitivity in the sense that it allows an assessment of whether the active treatments are effective. It does not, however, provide any information about whether a proportional active-placebo difference would be achieved with, for example, half or double the active-treatment dose and as such, appears to be suboptimal. The imposition of stringent acceptance criteria for equivalence may reduce the risk of falsely accepting as equivalent a nonequivalent T product. However, it does not correct for the inability to make a rigorous within-study assessment of sensitivity to differences in delivered dose.
With respect to other considerations (discussed below) such as the doses employed, dose level separation, study duration, and the population enrolled (including the use of a population enriched in “responders”), all should be subservient to the fundamental requirement for assay sensitivity.
Selection of doses to be studied
Given the requirement for assay sensitivity, a suboptimal dose, for example, 50 μg FP (one puff of a Reference MDI, which has more than one puff per the approved minimal therapeutic dose) may be considered acceptable as one of the dose levels in a PD efficacy study (although the dose should not be a noneffect dose in the model employed). Similarly, a supratherapeutic dose level of salmeterol may be considered acceptable in a PD safety study.(80,81)
With respect to dose level separation, the minimum separation required to distinguish doses is desirable. This will depend upon the model employed and the OIP under test, but doses separated by a factor of four may be required in many PD efficacy models, while a lesser dose multiple may suffice in classical PD safety models, at least for ICSs and β2-agonists.(80–83)
Finally, because the lowest marketed dose of most Reference OIPs lies near the plateau of the dose–response curve, development of a half-strength T, that is, a dose level below the minimum therapeutic dose approved for R, may increase the ability of the study to differentiate doses, improve assay sensitivity and better define the dose–response curve. This can increase the precision of the dose–response characterization and, in turn, decrease the width of the confidence interval of relative potency estimated using the Finney bioassay or Emax modeling allied to bootstrap statistical methods. The latter, which is not specifically recommended by EMA, is more suited for assessing dose–response curve and saturation of effect (Emax) following administration of active treatments (half-strength and full-strength) and placebo treatment. The development and use in clinical trials of half-strength presentations may be complicated, however, by several factors, such as unacceptable dose-to-dose variations in delivered dose when very small quantities of drugs are administered. Use of a lower strength dose therefore should probably be reserved for situations where marketed doses are so high on the dose–response curve that the PD equivalence study will be impractical without use of the lower dose strategy. There is no consensus on this approach at EMA, and a discussion with the appropriate regulatory agency is therefore recommended before proceeding.
Clinical study model
As for dose selection for an equivalence PD study, a clinical model that can differentiate formulations should be employed in preference to the one that is more relevant for clinical use but which is less discriminating between dose levels. In this respect, the development paradigm for a generic/alternate OIP is quite distinct from that for a novel inhaled drug.
Efficacy pharmacodynamics
Careful selection of an optimal outcome measure and the clinical study design in which it is measured, are critical to the success of an efficacy PD equivalence study. It is not sufficient just to identify significant differences between active doses and placebo. The selected clinical study model must be capable of identifying significant differences in responses to different active doses. Most often this is done using the Finney bioassay statistical methods (as recommended by the EMA guideline). The statistical power of an equivalence study using this method is a function of both the variability of the measured response (s) and the slope of the dose–response (b). These two factors do not function independently, but as a ratio (s/b).(84) The smaller the value of this ratio, the more powerful the study. Thus, the question of how steep is “steep enough” can only be meaningfully answered in the context of variability of the response measurement in the study.
Two study models have been successfully used for the PD assessment of short-acting beta agonist equivalence (Table 4): careful assessment of bronchodilation caused by the beta agonist,(85) and β-agonist-induced inhibition of bronchoprovocation with methacholine or histamine.(86–88) Although each of these models provides technical and logistical challenges, when done carefully, both can provide sufficient statistical power—that is, a dose–response that is “steep enough.” Assessment of long-acting beta agonists will likely be achievable with minor adjustments in these approaches, such as adjustment of the timing of response measurements to account for later occurrence of peak effects than is seen with short-acting β-agonists.
For inhaled corticosteroids, identification of a clinical study model that provides sufficient statistical power for a PD bioequivalence comparison has been much more problematic. Several outcome measures and associated study designs have been proposed, but it appears that very few of these studies have been used with success in PD efficacy equivalence studies.
Safety pharmacodynamics
Equivalent systemic exposure should be demonstrated through a PK safety study, if possible. If PK equivalence criteria are not met, then demonstration of comparable safety between T and R is required. The appropriate duration of a safety study should depend on the therapeutic class of the active substance, and should confer adequate sensitivity. However, there is no clear regulatory guidance regarding duration of assessment for single-dose and multidose studies, and dose levels (e.g., approved dose vs. supratherapeutic dose). The data should demonstrate noninferior safety of T versus R based on relevant changes in vital signs, cardiovascular assessments, biochemical parameters, and frequency of adverse events. (The EU guideline uses the term “no worse
When are pediatric PD studies required, and why?
There is a considerable ongoing debate as to pediatric requirements for generic OIPs, fuelled in part by the lack of definitive data to guide appropriate recommendations. The EMA guideline differs between adults and children in terms of acceptability of PK efficacy data (and consequently, in the requirements for PD efficacy studies). Specifically, in children, the guideline suggests that PK studies are not an acceptable surrogate to assess efficacy. However, there is no clear rationale to support this difference in approach to adults versus children. The guideline, therefore, advocates the provision of pediatric clinical data to support the approval of generic OIPs in pediatric patients in all but those (rare) cases where equivalence between T and R OIPs may be satisfactorily concluded on in vitro grounds alone.(3)
The EMA guideline highlights several differences in anatomy and physiology between adults and children, which may in turn influence drug delivery, such as differences in airway caliber, breathing patterns, airway resistance, and inspiratory flow profiles. The guideline concludes therefore that extrapolating data from adults to children may, as a result, be difficult. However, although the differences noted in the guideline between children and adults are indeed valid, it is pertinent to question the relevance of these differences to the generic OIP context, that is, if two OIPs are shown to be equivalent in adults (in either a PK and/or PD model), what mechanisms could lead to their lack of equivalence in children?
Metered-dose inhalers in pediatric PD studies
Differences in external pMDI design that might impact patient–device interaction are relatively limited. Different pMDIs generally differ only modestly in terms of mouthpiece shape and diameter. Conversely, different pMDIs may interact with a given spacer in different ways; hence, the relative deposition from two devices may differ when compared with and without a spacer. The former comparison is obviously of particular relevance to pediatric use, and spacer data should always be provided to support a generic pMDI submission for a product indicated for use in children. If equivalence cannot be inferred from an in vitro comparison, the PK and possibly PD studies with a spacer will be required, per the EMA guideline. In addition, if the Reference product was not authorized in children, a full clinical development program is required for the Test if it is intended for pediatric populations.
It is debatable whether a spacer study relevant to children could be performed in adults. Differences in plume geometry, which could theoretically alter the relationship between two pMDIs in adults versus children in the absence of a spacer, are not relevant when a spacer is involved. Conversely, lower tidal volumes in children may theoretically increase aerosol losses (via impaction, adsorption, sedimentation, and coagulation) prior to emptying of the spacer, although even young children using a large volume spacer should be able to inhale the spacer volume within 10 sec or less.(89) Overall, therefore, it is unclear whether the physiological differences between adults and children could alter the relationship between two pMDIs shown to be equivalent in adults both with and without a spacer. Several participants suggested that if PK equivalence was demonstrated for two pMDIs in adult studies both with and without a spacer, then clinical and PD pediatric studies were not needed. Unfortunately, definitive data are lacking, so no consensus recommendations could be made.
Dry-powder inhalers in pediatric PD studies
For DPIs, the situation appears more complex still. Available devices are far more varied than pMDIs in terms of external design and the patient–device interface.(90) Internal resistance and flow rate dependency may also vary considerably between devices.(91–94) With respect to the patient–device interface, it is plausible that the relative effects of two very different devices (e.g., Accuhaler and Turbuhaler) on oral geometry may differ; hence, drug deposition may differ depending on the population in which they are used, although definitive in vivo data in this respect are lacking.
Another scenario in which pediatric data are likely to be relevant, is where the relative in vitro performance of two DPIs varies across clinically relevant flow rate ranges. For example, a potentially important difference in performance between two devices may become apparent at lower flow rates applicable to young children but not at flow rates applicable to adults. An alternative to conducting studies to assess the impact of low flow rates on drug deposition/effect in pediatric subjects may be to use targeted low flow rates in adult subjects (controlled by using an inhalation profile recorder). Some Workshop participants suggested that with equivalent PK and in vitro data at low and high flow rates, further pediatric studies of DPIs should not be required (assuming no differences in the patient–device interface). However, there was no consensus on this issue. See also a discussion on pediatric PK studies in this article.
A further often cited consideration when discussing DPIs relates to the differences in operational handling between devices. Such differences may clearly be very relevant to the successful use of two different devices by children (and indeed by other patient subgroups). It is, however, debatable whether specific studies should be undertaken to compare drug deposition or PD effects when such devices are used in different populations under nonoptimal conditions, because the ability to optimally use a device should be an absolute prerequisite to a physician prescribing any device to a given patient.
In conclusion, although there is currently a lack of consensus on this issue and no relevant North American guidelines, there is a greater likelihood that additional PD data may be warranted in children during development of a generic DPI, although specific development requirements can only be validly assessed on a case-by-case basis. The greater the similarity between generic and reference DPIs, the less onerous requirements are likely to be.
Demonstrating Device Design Similarity
Device similarity and patient interface
The design of an OIP device and the interaction between the device and the patient are integral to reliable and consistent in vivo drug delivery. When changes in device design occur, these factors should be considered for demonstrating equivalence to the original device. Determining what changes in the device design are of practical and clinical relevance, can be difficult due to the lack of general IVIVCs for OIPs. It is a challenge to discern, a priori, design features that could impact patient handling and clinical outcomes. Published equivalence studies for pMDIs and DPIs do not typically contain extensive device details. As a result, the degree of device design difference that is permissible between OIPs or two versions of the same product is an area of considerable debate. The discussion is further complicated by the diversity of inhaled device designs and the multitude of potential changes between T and R or the changes that can occur during a product life cycle for a given OIP.
The importance of considering device design similarity in the context of equivalence comparisons of OIPs is recognized by regulatory authorities in Europe, Canada, and the United States, although their approaches and pathways to regulatory approval for device changes are different.(95) The EMA guideline states that the patient handling of T and R devices should be similar if equivalence is to be demonstrated by the in vitro route alone. In Canada, the regulations require that device design attributes be considered as they relate to functionality and potential impact on safety and efficacy. In the United States, where an aggregate “weight of evidence” approach to demonstrating BE has been recommended, it is recognized that similarity of device shape and equivalence in design and operating principles are expected to be important for patient use and therefore for the OIP overall equivalence.
Products showing equivalence under standardized in vitro test conditions or in a clinical testing environment, where device operation is closely monitored, may not necessarily demonstrate the same therapeutic outcome as in patients' hands outside the clinic. Variations in sound, taste, or other physical sensations associated with the OIP administration may impact patient's perceptions and, therefore, potentially their compliance with therapy. For example, upon the transition from CFC to HFA pMDIs, the “cold freon” sensation of CFC sprays disappeared due to the softer, warmer HFA sprays, and this was reported by the patients as “device malfunction.” Similarly, substitution of one DPI design for another can lead to patient mishandling if design features such as dose loading or mouthpiece geometry are modified from the original device.
Approaches to demonstrating similarity of OIP devices
The 2010 IPAC-RS Device Change Survey,(96) focusing on the design, material, and manufacturing changes, and the subsequent discussions at a breakout session of the 2010 ISAM/IPAC-RS Workshop(97) provided an opportunity to gather information on current attitudes and practices among pharmaceutical and device manufacturing industries, regulatory authorities, and academia with regard to OIP device changes and criteria for device similarity. The Workshop participants discussed the rationale and type of testing needed to determine equivalence when well-defined device changes occur (e.g., material changes, device shape). The objectives of the Workshop deliberations were to (1) describe current views regarding device changes between T and R or postapproval; (2) facilitate a move toward consensus on appropriateness and type of in vitro and in vivo testing for device changes, especially in light of risk management, quality-by-design (QbD), and self-regulation applied to device changes; and (3) highlight areas where regulatory requirements may differ from what is perceived to be technically required.
Both the results of the Device Change Survey and discussions at the Workshop indicated that current approaches to implementing and justifying OIP device changes are diverse and somewhat arbitrary, and that testing was often performed merely to meet perceived regulatory expectations and not technically warranted.
Significant themes that emerged included the need for regulatory interaction, incorporation of risk management, classification of OIP device changes, and better regulatory guidance or a best practice document prepared by the industry.
Interaction with regulatory agencies
Participants expressed a strong desire for further dialogue between industry and regulatory bodies to increase understanding of regulatory expectations related to OIP device changes. There is a striking lack of consistency in approaches to support device changes, as reflected in the IPAC-RS Device Change Survey results. Testing is often conducted not because of an assessment of the potential impact of a device change on the Critical Quality Attribute(s) of a product but because of perceived regulatory expectations. It was agreed that development of a rigorous, science-based approach may be needed to ensure that any critical impact(s) of a device design change could be assessed and addressed, and that increased self-regulation by manufacturers could make the development and approval of changed OIPs more efficient. Many participants also commented that it would be valuable to discuss specific challenges presented by OIPs with reviewers and regulatory agencies.
Use of risk management
Many Workshop participants had experience using risk management tools as a part of device development, and felt that these tools could play a greater role in the development of OIP devices specifically, although there was a lack of consensus on how they should be applied. Although these tools are based upon well-established medical device standards such as ISO 13485 & ISO 14971, participants were uncertain of how they could or should be used to manage OIP device changes; additionally, there was concern about the acceptability of these tools to regulatory agencies, because most OIPs are regulated as drugs, not medical devices.
The IPAC-RS Device Change Survey results and the Workshop discussion also suggested that there was an interest from industry in moving toward a QbD approach as the standard way of constructing regulatory submissions for OIPs. This approach is closely related to the Risk Management and Quality System approach that exists for Medical Devices in Europe and North America, and many contributors felt it would provide a platform to deal with future changes in a more scientific and pragmatic manner. Due to a lack of clarity about how this might work, however, there was some hesitancy to pursue a QbD path. Although many companies were using risk management tools in development of devices and assessment of changes, few were including such information in regulatory submissions (e.g., New Drug Applications, Marketing Authorization Applications), thereby using an opportunity to challenge the current paradigm. Industry experience with regulatory responses to QbD submissions for OIP devices is therefore limited.
Classification of device changes, guidance, and best practice
Developing a prescriptive guidance document addressing all types of device changes would be extremely difficult and of limited value due to the vast range of device technology that exists now and may emerge in the future. However, many Workshop participants felt that additional guidance focused on risk management and risk assessment of device changes would be of value. The ISO 20072 standard for aerosol devices was acknowledged as an example of a helpful guidance document that ensures that the critical aspects of OIPs are adequately considered, but places the majority of responsibility for the risk assessment and decision making with the manufacturer.
A guidance for device change management need not come from regulators. The participants suggested that industry could develop a document describing best practices, points to consider and consensus recommendations.
Some classification of device changes that would guide manufacturers' efforts should, however, be either issued or endorsed/accepted by regulators to be of maximum value.
Recommendations from Discussions on Device Similarity
A variety of routes exist for achieving consensus on testing to determine bioequivalence of changed devices. Increased alignment within and between all stakeholders, including industry and suppliers, as well as regulatory and standard-setting bodies such as the International Organization for Standardization (ISO), the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and the Global Harmonization Task Force (GHTF), would contribute to a more harmonized, consistent and transparent approach.(98) Enhanced communication between and within regulatory agencies, such as between the FDA Center for Devices and Radiological Health (CDRH) and the FDA Center for Drug Evaluation and Research (CDER) could also facilitate further alignment.(99)
Regulatory or “best industry practices” guidelines should be developed or refined to be (1) flexible rather than prescriptive; (2) based on risk management principles, such as per ISO 14971 and ICH Q9; (3) aiming to classify and consequently qualify a device change based on an assessment of its potential impact on critical quality attributes; (4) incorporating appropriate technical input; and (5) linking testing requirements to clinically relevant parameters.
These considerations should be taken up jointly by industry and regulatory bodies. Prospectively, these stakeholders should assess whether a classification system for OINDP device changes would help define the testing approach and regulatory pathway to support these changes and, if it would, how OIP device changes could be categorized, as well as who should decide when a scientific and technical justification for a change is adequate and acceptable. Additionally, stakeholders should consider what benefits exist to increased self-regulation of activities by industry, based on risk management and quality management systems (as practiced for non-OIP medical devices). Considerations for increasing industry self-regulation include: what would facilitate or hinder this process, what steps would be necessary for implementation of such a system, and what would be required for its recognition by regulators. Among potential benefits of increased self-regulation are improved and more consistent OIP performance, more robust quality systems, and better transparency.
Areas for Further Work and Discussion
This report summarized the regulatory approaches, state of knowledge, and current understanding regarding equivalence determinations for locally acting orally inhaled products, as discussed at the 2010 ISAM/IPAC-RS European Workshop. The consensus positions were presented and discussed in the four major sections of this report (in vitro, PK, PD, and device similarity). Even though generally consistent recommendations exist in this field (albeit with some differences in the details of regulatory requirements among the European Union, Canada, and the United States), there remain areas with insufficient knowledge and therefore in need of further research and discussion. These open-question areas are listed in Table 6.
The ISAM/IPAC-RS Workshop participants expressed the hope that future research by interested stakeholders, and constructive discussions in public fora would address these open questions.
Footnotes
Acknowledgments
This article represents a report of the workshop's discussions, and may not fully reflect the positions of individual authors or of any organization with which the authors are affiliated. The authors are grateful to all speakers and attendees of the ISAM/IPAC-RS Workshop, as well as to ISAM and IPAC-RS organizations for the support of this project. The authors also thank Julianne Berry (Merck), a member of the ISAM/IPAC-RS Workshop Organizing Committee, and ISAM/IPAC-RS Writing Committee, for her valuable contributions and helpful discussions.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.