
Abstract
Abstract
Background:
The long-term safety of a new asthma therapy combining fluticasone propionate and formoterol fumarate (fluticasone/formoterol; flutiform®) was assessed.
Method:
In an open-label study, mild to moderate-severe asthmatics (≥12 years; N=472) were treated twice daily with fluticasone/formoterol 100/10 μg (n=224) or 250/10 μg (n=248) for 6 months (n=256) or 12 months (n=216). The primary and secondary objectives were the long-term safety and efficacy of fluticasone/formoterol, respectively.
Results:
In total, 413 (87.5%) patients completed the study (of which 175 participated for 12 months). Adverse events (AEs) were reported by 174 patients (36.9%): 67 (29.9%) in the 100/10 μg group and 107 (43.1%) in the 250/10 μg group. The most common AEs (>2%) were nasopharyngitis, dyspnea, pharyngitis, and headache; the majority were mild to moderate. Only 18 (3.8%) patients reported AEs considered study drug–related. Five patients per group experienced 12 serious AEs; none was study medication–related. Asthma exacerbations were reported by 53 patients (11.2%): 46 mild to moderate and nine severe. Clinical laboratory tests and vital signs showed no abnormal trends or clinically important or dose-response–related changes. The efficacy analyses showed statistically significant improvements at every time point throughout the study period at both doses.
Conclusion:
Fluticasone/formoterol had a good safety and efficacy profile over the 6- and 12-month study periods.
Introduction
Despite significant advances in asthma management,16–21 poor control of symptoms and exacerbations persist for a substantial proportion of patients. This may be a consequence of inadequate drug delivery due to poor inhaler technique, the choice of asthma therapy, or other factors such as lack of patient adherence to prescribed treatment.1,22–27 The capacity for therapies to reach the small airways, which are an important site of inflammation in asthma, may also affect treatment efficacy.28,29 However, the clinical significance of ICS/LABA deposition in the distal airways has not yet been demonstrated.30,31
The ICS, fluticasone propionate (fluticasone), delivers a potent and sustained anti-inflammatory effect.17,32–34 The LABA, formoterol fumarate (formoterol), has a fast onset of action and a prolonged bronchodilatory effect.5,35–38
Fluticasone and formoterol have now been combined in a single aerosol inhaler (fluticasone/formoterol; flutiform®). This study assessed the long-term safety and efficacy of this new combination aerosol in adolescents and adults with mild to moderate-severe asthma.'
Materials and Methods
Study design
This was a long-term (up to 12 months), open-label study assessing the safety and efficacy of fluticasone/formoterol 100/10 μg and 250/10 μg, administered twice daily (b.i.d.). The study was conducted in five European countries (Germany, Hungary, Poland, Romania, United Kingdom; EudraCT number: 2005-003518-14; US NCT number: NCT00394121), in accordance with ICH Good Clinical Practice (GCP), the Declaration of Helsinki, the European Union Clinical Trials Directive (2001/20/EC), the GCP Directive (2005/28/EC), and the prevailing local laws and customs of the participating countries. Independent Ethics Committees at each center reviewed and approved the protocol, and written informed consent was obtained from all patients, or the parents or legal guardians of those patients below 18 years of age, prior to screening.
Patients
Adults and adolescents (≥12 years) with mild to moderate-severe asthma were eligible for enrollment if they had a history of asthma for ≥12 months 39 and a documented use of ICS asthma maintenance therapy for ≥4 weeks prior to screening at a dose not greater than 500 μg/day inhaled fluticasone or equivalent ICS. Eligible patients had to demonstrate a forced expiratory volume in the first second (FEV1) of between 40% and 85% (inclusive) of predicted normal values at both screening and baseline (week 0), following appropriate withholding of asthma medication. At screening, patients were required to discontinue any LABA treatment at least 24 hr before the visit for pulmonary function testing and not use a short-acting bronchodilator as a rescue medication within 6 hr prior to the visit. At baseline, patients were not permitted to take fluticasone or rescue medication (salbutamol) within 12 hr or 6 hr, respectively, before the visit. In addition, patients had to show a documented reversibility of ≥15% in FEV1 within 6 months of screening. During the 14±3 day run-in period, patients were administered fluticasone by hydrofluoroalkane (HFA) pressurized metered-dose inhaler (pMDI) b.i.d. and had to use ≥2 inhalations/day of rescue salbutamol for ≥3 days, as well as experience ≥1 night with sleep disturbance or ≥3 days with asthma symptoms.
Exclusion criteria included life-threatening asthma within 12 months, systemic (oral or injectable) corticosteroid medication within 3 months, omalizumab within 6 months, or leukotriene receptor antagonist within a week prior to screening. Patients with an upper or lower respiratory tract infection within 4 weeks prior to screening or during the run-in, significant, non-reversible pulmonary disease, a smoking history (equivalent to 10 pack years, or had smoked within 12 months), or receiving β-blocking agents, tricyclic antidepressants, monoamine oxidase inhibitors, astemizole, quinidine-type antiarrhythmics, or potent CYP3A4 inhibitors in the week prior to the screening visit were also excluded.
During the treatment period, fluticasone/formoterol was administered for either 6 months or 12 months. The treatment period comprised scheduled visits at weeks 2 and 4, and monthly thereafter. Patients were given a diary card and a peak flow meter and, during the run-in, entered information into an Interactive Voice Response System diary. During both the run-in and treatment periods, patients recorded their pre-dose peak expiratory flow rate (PEFR) twice daily, their daily assessment of asthma symptoms, and daily rescue medication use.
Interventions
During the 14±3 day run-in period, patients were assigned to one of two groups (Fig. 1) and self-administered fluticasone (suspension delivered by HFA pMDI) at a dose dependent on their steroid use prior to screening: those requiring 100–249 μg/day fluticasone (or equivalent) were assigned 100 μg/day fluticasone (one actuation 50 μg b.i.d.), whereas those using 250–500 μg/day fluticasone (or equivalent) received 250 μg/day fluticasone (one actuation 125 μg b.i.d.).
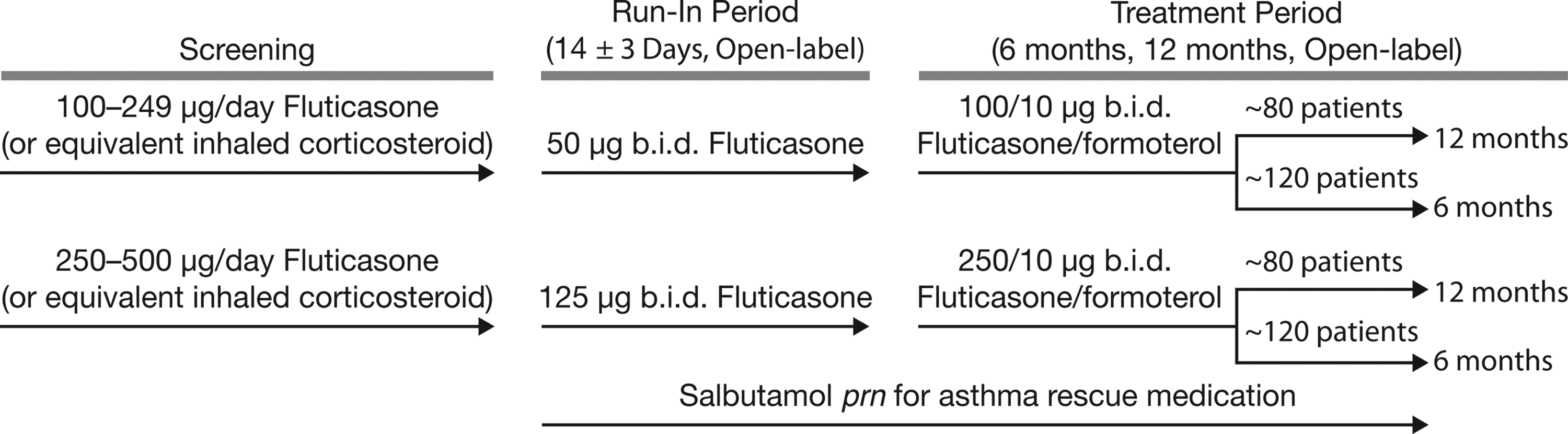
Study diagram. prn, as needed.
During the treatment period, patients received either fluticasone/formoterol 100/10 μg b.i.d. (HFA suspension; two actuations 50/5 μg, b.i.d.) or 250/10 μg b.i.d. (HFA suspension; two actuations 125/5 μg, b.i.d.), depending on whether they had 100 μg/day or 250 μg/day ICS during the run-in period. Study medications were administered via a pMDI without the use of a spacer, and patients were instructed to take morning and evening doses timed apart as evenly as possible. Patients were also required to leave a 1-min interval between inhalations and wash their mouth thoroughly after dosing. Rescue salbutamol (100 μg/actuation) was available, as needed, throughout both the run-in and treatment periods.
Safety assessments
Safety assessments included medical history at screening, physical examinations at screening and baseline, and vital sign assessments prior to pulmonary function tests (PFTs) and within 30 min before the morning dose of study medication. A safety follow-up, by telephone, took place 2 weeks after the last dose of treatment. A 12-lead electrocardiogram (ECG) was performed prior to PFTs at screening and before morning dosing at each study visit; clinical laboratory tests were performed at screening, weeks 0 and 4, and months 3, 6, and 12 or at the final visit for patients who discontinued prematurely. Adverse events (AEs), their severity, outcome, and relationship to study medication, and the proportion of patients who were withdrawn because of AEs were recorded throughout the study period.
Patients were withdrawn if they experienced severe asthma exacerbations requiring medical intervention. A mild-to-moderate asthma exacerbation was defined as night awakenings due to asthma on 2 or more consecutive days, or the additional use of rescue medication of ≥3 inhalations/day with respect to baseline on 2 or more consecutive days, or morning pre-dose PEFR >30% below baseline values on at least 2 consecutive days. The baseline values for the run-in period were defined as the PEFR measured at screening; for the treatment period, this was defined as the average of three morning PEFR measurements taken at baseline. A severe asthma exacerbation was defined as the deterioration in asthma symptoms requiring additional therapy, e.g., a systemic steroid, a visit to the emergency room, or hospitalization due to asthma.
Patients were also withdrawn if their pre-bronchodilatory FEV1 decreased to <40% of predicted normal values, prebronchodilator PEFR decreased to ≤75% of baseline average values on 4 consecutive days, if they used ≥12 actuations/day of rescue salbutamol on ≥3 days/week, recorded a sleep disturbance score of ≥3 on three consecutive nights, used prohibited medications for asthma exacerbations, or experienced a serious or unexpected AE. Patients who withdrew from the study due to an AE were followed up with appropriate clinical and/or laboratory tests until satisfactory resolution of the AE.
Patients returned all used and unused study drug to the site at each visit. During the run-in and treatment periods, patients recorded the number of inhalations of fluticasone and the time and number of inhalations of study drug, respectively. At each study visit, the dose counters on the fluticasone/formoterol inhalers were checked to confirm the number of actuations taken. Patients with <70% adherence to study medication were withdrawn from the study.
Efficacy assessments
Efficacy assessments were the secondary endpoints of this study and included the spirometry performed at screening, within 30 min before morning dosing, and 1 hr after dosing at each visit during the treatment period. LABA medication was discontinued at least 24 hr and short-acting β2-agonist medication within 6 hr prior to the first spirometry at screening, and the use of rescue salbutamol within 6 hr of all scheduled PFTs. All PFTs were carried out using a spirometer 40 and included the forced vital capacity (FVC) (the maximum volume of air expired as forcefully and as rapidly as possible after maximum inspiration), PEFR (the maximum flow rate obtained during the FVC procedure), FEV1, and FEV1 % predicted based on the patient's age, height, and gender, determined according to guidelines for adults (≥17 years) 41 and for adolescents (12–16 years). 42
During the study, patients recorded their daily morning and evening pre-dose PEFR, the time and number of inhalations of both study and rescue medication (patients were to contact the study site if they had taken 12 or more inhalations of rescue medication on any one day), asthma symptom scores [from 0 (no symptoms) to 5 (asthma was so severe that the patient was unable to go to work or school or carry out normal daily activities)], and sleep disturbance scores [from 0 (patient slept through the night and experienced no asthma) to 4 (patient could not sleep at all because of asthma)].
Statistical analyses
No statistical analyses were performed on the safety parameters.
Statistical analyses for FEV1, FVC, and PEFR and the changes from baseline to each subsequent visit and within each dose group were analyzed using a paired t-test. All statistical analyses were performed at the 0.05 significance level unless otherwise noted.
The safety population comprised all enrolled patients who had at least one inhalation of study medication; the full analysis set (FAS) included patients in the safety population with at least one efficacy measurement at baseline and post-baseline, and the per-protocol (PP) population comprised patients in the FAS without a major protocol violation. Sample size estimates were not formally calculated but were based on the ICH guidelines on the minimum number of patients required for the long-term treatment of non-critical conditions.
Results
Overall, 472 patients entered the treatment period and 413 completed the study (Fig. 2). The safety population included all 472 patients, the FAS contained 466 patients (221 in the 100/10 μg b.i.d. and 245 in the 250/10 μg b.i.d. group), and the PP population included 390 patients. Overall, 77 (16.3%) patients had a major protocol violation [31 (13.8%) in the 100/10 μg b.i.d. group; 46 (18.5%) in the 250/10 μg b.i.d. group], including violations of the inclusion/exclusion criteria [51 patients (10.8%)], non-adherence to study medication [23 patients (4.9%)], the use of prohibited concomitant medications [4 patients (0.8%)], or failure to withhold specific medications prior to PFTs [1 patient (0.2%)].
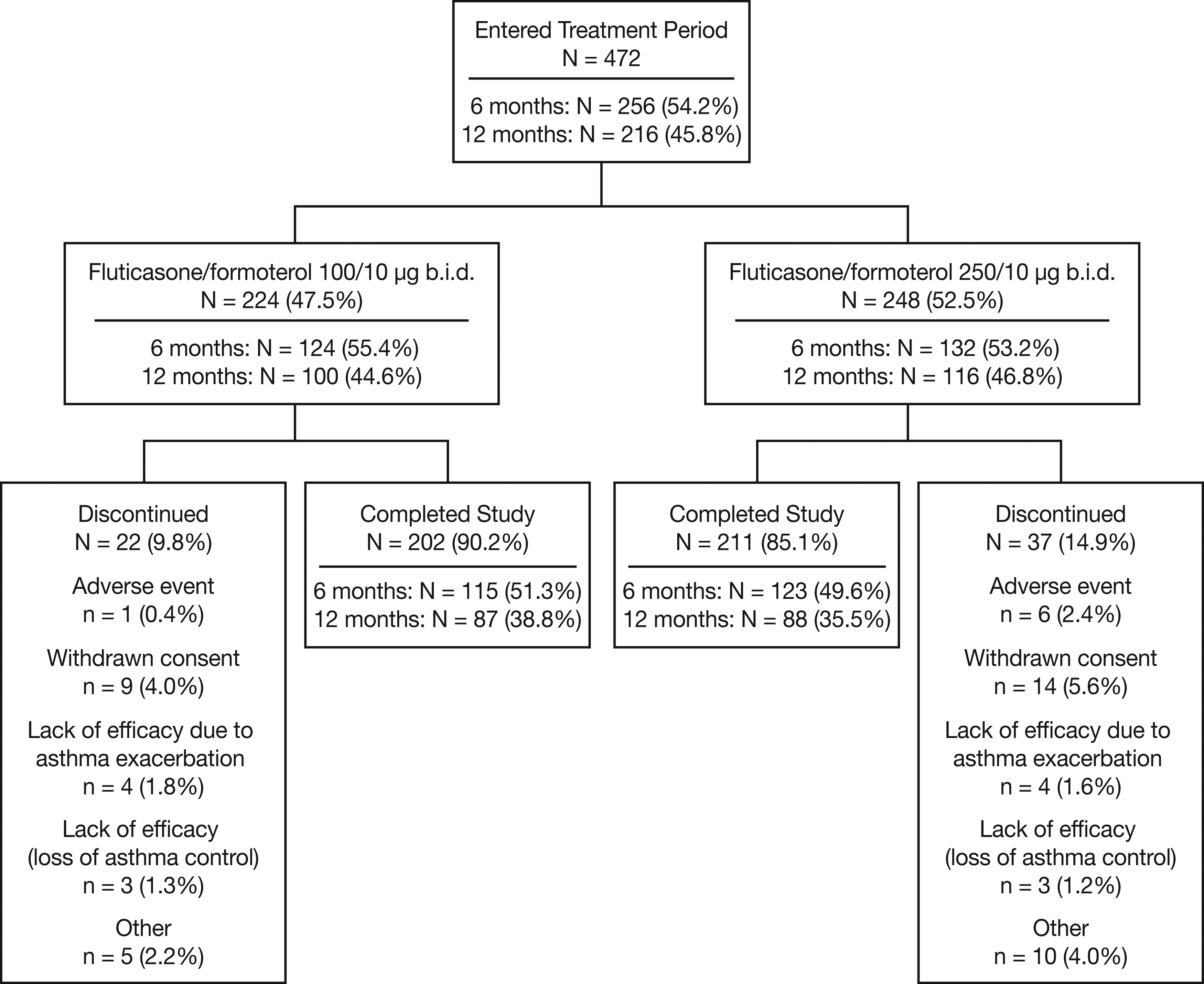
Patient flow diagram.
In general, the two treatment groups were well matched with respect to baseline characteristics (Table 1). The 250/10 μg b.i.d. treatment arm had a ≥5% higher incidence of patients with a medical history that included abnormalities in the cardiovascular and genitourinary systems, and the 100/10 μg b.i.d. group had a higher proportion of patients in the 12–17-year age group compared with the 250/10 μg b.i.d. group (17.9% vs. 6.5%).
N, number of patients in a treatment group; n, number of patients with data available; SD, standard deviation; b.i.d., twice daily.
N=213 for weight, bN=232 for weight, cN=445 for weight.
FEV1 % predicted and FEV1 are from baseline (week 0), and reversibility is from screening.
N=216 for reversibility, fN=231 for reversibility, gN=447 for reversibility.
Safety and tolerability
The duration of exposure to study medication is presented in Table 2.
N, number of patients in a treatment group; SD, standard deviation; b.i.d., twice daily.
Overall, 174 (36.9%) patients reported AEs (Table 3). The majority were mild to moderate in severity; those occurring with an incidence of >1% are presented in Table 4. The numbers of patients reporting severe AEs are displayed in Table 5; asthma was the only severe AE reported by more than one patient. In total, 18 (3.8%) patients reported AEs possibly or probably related to study medication, including asthma (n=2) and dysphonia (n=5) reported for more than one patient.
N, number of patients in a treatment group; b.i.d., twice daily.
Investigator assessment was possibly or probably related to study drug.
N, number of patients in a treatment group; b.i.d., twice daily.
An asthma exacerbation was considered an adverse event if it did not resolve with the study drug treatments (including salbutamol) and additional medication was required (e.g., systemic glucocorticosteroids).
N, number of patients in a treatment group; b.i.d., twice daily.
An asthma exacerbation was considered an adverse event if it did not resolve with the study drug treatments (including salbutamol) and additional medication was required (e.g., systemic glucocorticosteroids).
No deaths were reported. Five patients in each treatment group experienced a total of 12 serious AEs, all deemed either not or unlikely to be study medication–related. Three patients (1.3%) receiving 100/10 μg b.i.d. and 11 (4.4%) receiving 250/10 μg b.i.d. fluticasone/formoterol discontinued from the study, at least in part, due to an AE; two patients who received 100/10 μg b.i.d. had an asthma exacerbation and anxiety-induced respiratory syndrome, respectively; six patients who received 250/10 μg b.i.d. experienced severe dyspnea during activity, worsening of asthma, a serious asthma exacerbation, increased breathlessness, increased asthma symptoms, and upper respiratory tract infection, respectively.
Six patients had laboratory abnormalities that were mild to moderate, non-serious AEs: one patient each had increased white blood cell and neutrophil counts, increased white blood cell count, increased platelet count, increased aspartate aminotransferase (AST) and alanine aminotransferase, increased AST, and increased sodium. Four patients had elevated glucose values that met the criteria for potential clinical significance, three of whom had a history of diabetes, and all had high plasma glucose levels at each visit, including at screening and at baseline.
Three patients reported AEs related to increased blood pressure: two in the 250/10 μg b.i.d. group reported worsening hypertension (not study medication–related), and one in the 100/10 μg b.i.d. group reported arterial hypertension (possibly study medication–related). All AEs were assessed as mild to moderate in severity and resolved or stabilized with medication.
Four patients had one post-baseline ECG QTcF interval that was >500 msec, but continued on the study medication with no subsequent QTcF intervals of >500 msec and without cardiovascular or dysrhythmia events associated with the QT prolongation. Overall, clinical laboratory tests, vital sign evaluations, and ECGs showed no abnormal trends or dose-response–related changes.
Asthma exacerbations
In total, 53 (11.2%) patients reported asthma exacerbations, including 46 (9.7%) reporting mild to moderate exacerbations [22 (9.8%) in the 100/10 μg b.i.d. and 24 (9.7%) in the 250/10 μg b.i.d. group] and 9 reporting severe exacerbations [1.9%; 3 (1.3%) in the 100/10 μg b.i.d. and 6 (2.4%) in the 250/10 μg b.i.d. group]. One patient administered 100/10 μg b.i.d. and 3 administered 250/10 μg b.i.d. discontinued prematurely because of asthma, including one as a result of serious asthma exacerbation.
Efficacy
Efficacy analyses were carried out on the FAS and showed statistically significant improvements overall and for both treatment groups for each efficacy assessment (Supplementary Tables S1–S4; Supplementary Data are available online at www.liebertonline.com/jamp). The data shown in Figure 3 illustrate the time course of pooled efficacy results for all patients that reached month 12. Data show that, within the first 2 months of treatment, overall efficacy parameters improved and that good results were sustained throughout the 12-month treatment period.

Summary of efficacy endpoints for patients that reached month 12. Mean pre-dose FEV1 (L) and PEFR (L/min) values from day 0 to month 12 are shown.
Discussion
Fluticasone/formoterol combination therapy demonstrated a good safety and tolerability profile over the 12-month study period. Altogether, 174 patients (36.9%) reported AEs, including 67 (29.9%) in the 100/10 μg b.i.d. and 107 (43.1%) in the 250/10 μg b.i.d. group, with the majority either mild or moderate in severity. The incidence of AEs observed with fluticasone/formoterol in the present study is in line with that observed in previous long-term studies of ICS/LABA combinations.43,44 For example, the overall incidence of AEs was 48.6% with fluticasone propionate/salmeterol xinafoate (250/50 μg b.i.d.) and 52.3% with budesonide/formoterol fumarate (200/6 μg once daily or 200/6–400/12 μg b.i.d.) after 1 year's treatment in adults with persistent asthma. 43 Thus, the rates of AEs reported here do not appear to be unusual for combination therapy administered over a period of up to 1 year.
The AE profile observed with the fluticasone/formoterol combination is consistent with that of the individual component drugs and in line with those reported for other ICS/LABA combinations. The most common AEs observed with fluticasone/salmeterol therapy in a 1-year study were nasopharyngitis (13%), upper respiratory tract infection (13%), and headache, sinusitis, and influenza (each 5%)19,45; the type and incidence of AEs were similar to those for fluticasone alone. The most common treatment-related AEs reported in previous studies included headache (incidence 1–5%), throat irritation/cough (1–5%), hoarseness/dysphonia (2–7%), and oropharyngeal candidiasis (1–5%). 46 With the budesonide/formoterol combination, the most common AEs observed in a 6-month study were respiratory infection (36%), viral infection (10%), bronchitis, pharyngitis, and headache (each 6%), and sinusitis, rhinitis, and dysphonia (each 5%). 47 Common treatment-related events reported with beclometasone/formoterol in a 24-week study include headache, hoarseness, and pharyngitis. 48 Moreover, comparison studies suggest no significant differences in AE profile between the different ICS/LABA combinations43,44,49,50 or between ICS/LABA combinations and ICS therapy alone.51,52
In the current study, nasal or oropharyngeal AEs made up a notable proportion of the AEs. This finding is not unexpected given the known oropharyngeal effects of ICS therapy.53,54 In the study, the majority (>75%) of these events were mild in severity, none of the events were severe, and most were not considered related to study medication.
Patients administered fluticasone/formoterol 100/10 μg b.i.d. reported fewer AEs related to the respiratory system compared with those administered 250/10 μg b.i.d., which may be reflecting differences in the underlying asthma severity between the two groups. The patients assigned to this treatment group were already receiving 250–500 μg/day fluticasone (or equivalent) prior to study enrollment compared with those in the 100/10 μg b.i.d. group who were administered 100–249 μg/day fluticasone (or equivalent). The greater incidence of dysphonia (2.4% vs. 0.4%), for example, may have been a consequence of the increased exposure of patients to higher doses of ICS in the 250/10 μg b.i.d. group compared with the 100/10 μg b.i.d. treatment arm 55 and may also have been affected by the fact that spacers were not used in this study. The laryngeal deposition of the ICS has been reported to cause the myopathy of the arytenoid muscles and dysphonia, which seems to be dose-related. 55
No deaths were reported, and the 12 serious AEs experienced by the 10 (2.12%) patients were considered not related (n=9) or unlikely related (n=3) to study medication, including three that were respiratory system–related (pneumonia, anxiety-induced respiratory syndrome, and asthma exacerbation).
Asthma exacerbations, principally mild to moderate, were reported by 53 (11.2%) patients; only 9 (1.9%) experienced severe exacerbations, with the incidence of severe exacerbations ∼1% higher in the 250/10 μg b.i.d. than in the 100/10 μg b.i.d. group (2.4% vs. 1.3%, respectively). It could be concluded that this is possibly a result of the severity of the underlying disease in the group receiving the higher dose of combination therapy.
The clinical assessments and vital signs showed no significant or abnormal trends or dose-response changes in either treatment group over the 6–12-month period.
The secondary endpoints successfully demonstrated the efficacy of the combination therapy at both dose strengths. Clinically important improvements were seen at the α=0.001 level for mean changes in the pre-dose as well as in the predose to 1-hr post-dose assessments of FEV1, FEV1 % predicted, PEFR, and FVC measurements: improvements were observed within a short time period, at each assessment time point, throughout the treatment period, and for both treatment arms and overall.
As with all chronic diseases, patient adherence to study medication is an important factor.55–58 Patients in this study demonstrated good adherence to therapy in both dosage groups, with only 23 (4.9%) considered protocol violators because of non-compliance: this is especially noteworthy considering the study period (up to 12 months). The fact that patients underwent monthly visits in a clinical trial setting may have had a positive effect on adherence to study treatment. However, the rapid bronchodilation reported with fluticasone/formoterol therapy, reflecting the rapid effect of the formoterol component of the combination, 59 could also be an important factor in improving treatment adherence. This notion is supported by the fact that non-adherent patients would improve their adherence if they were able to feel a therapy working soon after taking it.60,61
The safety and efficacy of both fluticasone and formoterol are extensively documented in the literature.5,17,32–38 The administration of fluticasone/formoterol from a single pMDI allows patients to experience rapid bronchodilation after dosing while providing sustained anti-inflammatory effects, via a commonly used and familiar device. 26 Together, these factors may provide incentives for patients to adhere to their treatment regimen and achieve better asthma control.
In conclusion, this study successfully demonstrated the long-term safety and efficacy of the new fluticasone/formoterol combination therapy for patients whose asthma is not adequately controlled with ICS monotherapy or is already controlled with an ICS/LABA combination.
Footnotes
Acknowledgments
The authors thank all study investigators and participants, the study team at SkyePharma, colleagues at Mundipharma Research Limited (MRL), including Dr. Evelin Kozma, and Oxford PharmaGenesis™ Ltd. for medical writing assistance on behalf of MRL. The study was sponsored by SkyePharma, Switzerland. Flutiform® is a registered trademark of Jagotec AG and is used under licence. Clinical trial registration numbers are: EudraCT number: 2005-003518-14; US NCT number: NCT00394121.
Author Disclosure Statement
Adel H. Mansur took part in this clinical trial and has received honoraria and consulting fees from the sponsor. Kirsten Kaiser is an employee of SkyePharma (the study sponsor).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.