
Abstract
Abstract
This review article discusses the development of respiratory therapeutics containing bacteriophages indicated for lung infections, specifically those that have become increasingly difficult to treat because of antibiotic resistance. Recent achievements and remaining problems are presented for each step necessary to develop a bacteriophage-containing dosage form for respiratory drug delivery, including selection of appropriate bacteriophages for therapy, processing and purification of phage preparations, formulation into a stable, solid dosage form, and delivery device selection. Safety and efficacy studies in animals and human subjects are also reviewed.
Introduction
Antibiotics can be categorized based on their specificity. Broad-spectrum antibiotics can be used against a wide range of bacteria, but even narrow-spectrum antibiotics typically target a whole class of bacteria, such as Gram-negative or Gram-positive. Due to the relatively nonspecific mechanisms of action of some broad-spectrum antibiotics, bacterial strains are often not identified before therapy is initiated. However, the nonspecific activity also results in some of the commonly observed antibiotic side effects, e.g., diarrhea, associated with the disruption of beneficial human microflora. Arguably the greatest weakness of antibiotics is that their effectiveness is diminished by the emergence of bacterial resistance. Within two decades after the discovery of penicillin, it was recognized that bacterial resistance would impact the efficacy of antibiotics. Sir Alexander Fleming, among others, warned early—and to no avail—against improper use of penicillin, e.g., using insufficient doses or terminating treatment prematurely. The emergence of resistant bacterial strains has been accelerated by a variety of factors, such as the overuse and misuse of antibiotics in clinical practice and in patients' hands, and indiscriminate application of antibiotics in animal husbandry and fish farming. It has typically taken between 3 and 30 years following the introduction of a new antibiotic to observe the onset of resistance.(2)
Bacterial resistance has become a significant problem in the treatment of infections of the respiratory system. Refractory or chronic infections are common in patients with compromised lungs, such as cystic fibrosis (CF) patients. Their lungs are colonized by a variety of bacteria, often initially by Staphylococcus aureus or Haemophilus influenzae and later typically by bacteria of the Burkholderia cepacia complex (Bcc) or by Pseudomonas aeruginosa.(3) Many of these bacteria have high intrinsic antimicrobial resistance or have acquired mutations that result in resistance to a broad range of antibiotics.(4,5) Similarly, highly resistant bacterial strains of Klebsiella pneumoniae, Streptococcus pneumoniae,(6) and Enterobacter spp. are encountered in hospital emergency care units and are a leading cause of mortality due to nosocomial pneumonia. Some of these bacterial species have now become resistant to all carbapenem antibiotics(7) via the transfer of genes that encode a highly effective β-lactamase enzyme.(8) In addition, it is likely that at least 5% of the more than 400,000 multidrug-resistant tuberculosis cases per year worldwide are caused by extensively drug-resistant Mycobacterium tuberculosis,(9) which is practically untreatable in developing countries.(10) Some of this high-level antibiotic resistance is attributable to the fact that several pulmonary pathogens are capable of adopting a biofilm lifestyle,(11) in which the bacteria are encased in a self-synthesized polymeric matrix that serves to protect them from antibiotic activity and immune responses. The exact mechanisms producing biofilm resistance remain undiscovered, but may be through a combination of bacterial adaptations, the net effect being that biofilm bacteria are nearly impossible to clear even using combinations of conventional chemical antibiotics.
More than 20 classes of antibiotics were introduced for clinical use between 1940 and 1962. However, only two have been added after this time, and very few are currently in development. Given the recent increase in the use and overuse of antibiotics, it is not surprising that now many bacterial species have developed resistance against more than one antibiotic. A recent study by Coates et al. estimates that 20 new classes of antibiotics would have to be discovered and developed in the next five decades to control bacterial infections at the current level.(12) Because it appears unlikely that this goal can be accomplished, even if stronger economic incentives for the development of new antibiotics were put in place, a search for alternatives to antibiotics seems warranted.
One alternative, phage therapy, has received renewed attention.(13,14) Bacteriophages, or phages, are viruses that infect and kill bacteria.(15) Figure 1 gives an overview of some selected milestones in the discovery of bacteriophages and their application to the treatment of respiratory infections. Phages were discovered independently by Frederick Twort and Felix d'Herelle at the beginning of the 20th century. D'Herelle soon thereafter initiated experimental treatment of bacterial infections with phage “cocktails” (preparations consisting of a collection of different phages active against multiple bacterial strains or species), despite the fact that little was known about phage particles at the time.(15,16) Early phage therapy was quite successful, leading to the development of many commercial phage preparations in the 1920s and 1930s by companies such as Eli Lilly & Company and widespread use of phage therapy in Europe and the United States. However, early clinical trials also suffered from poorly characterized phage preparations and a lack of understanding of phage biology. As a result, the efficacy of phage treatment was called into question, and phage therapy in the West was largely abandoned with the advent of chemical antibiotics.(17) Nevertheless, phage research intensified in the 1940s as phages were recognized by Max Delbrück, among others, as powerful tools for the exploration of genetics. The American Phage Group, coordinated by Delbrück, contributed largely to the scientific understanding of phages that emerged in the 1940s and 1950s. In 1969, the Nobel Prize in Physiology or Medicine was awarded to Max Delbrück, Alfred D. Hershey, and Salvador E. Luria, “for their discoveries concerning the replication mechanism and the genetic structure of viruses.”(18) Phage research became an exact science with rigorous quantitative methods. Some of the earlier failures of phage therapy could now be explained. It was realized that phages typically have a narrow bacterial host range, and that this host range specificity depends on an explicit match between the bacteriophage and bacterial surfaces or surface structures.
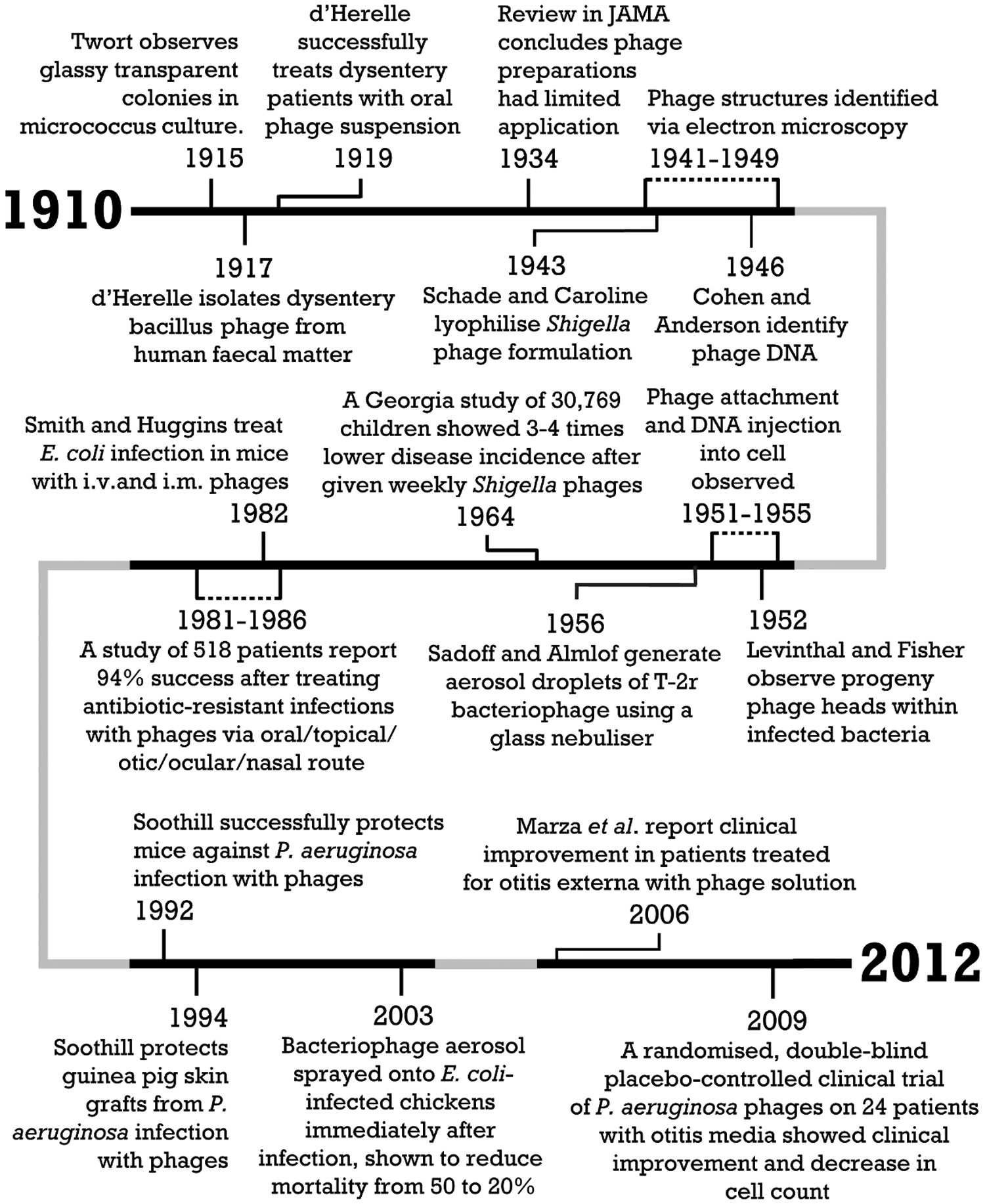
A historical overview of respiratory phage therapy.
The first reports of successful inhalation therapy using phages emerged around 1960.(19,20) Table 1 outlines selected studies of inhalation phage therapy in humans. Delacoste successfully treated patients with refractory cough using a nebulized phage cocktail.(19) Hoeflmayr nebulized a phage cocktail concomitantly with antimicrobial agents.(20) Endotoxins were intentionally left in the preparation to stimulate an immune response in the patients. The treatment was successful in 90% of patients with chronic bronchitis. In Eastern Europe, in particular in Russia, Poland, and Georgia, phage therapy has been continuously applied and further developed since the 1920s.(21) Phage therapy research centers include the Hirszfeld Institute of Immunology and Experimental Therapy(22) in Wrocław, Poland, and the Eliava Institute(23) in Tbilisi, Georgia. In Georgia, phage therapy is an accepted and widely used mode of treatment. Commercial phage products, including phage cocktails that are available to the public without prescription, are manufactured and approved in Georgia and Russia.(21) The prospects for respirable phages as an alternative to antibiotics appear promising.
In this article, we provide an overview of the individual steps that need to be taken to develop a bacteriophage dosage form for administration to the respiratory system. The first sections review the selection of adequate phages, their amplification, and purification. Then recent advances in the formulation of phage preparations into stable, respirable dosage forms are discussed and delivery device options are reviewed. Finally, the efficacy and safety of phage therapy as established by recent clinical trials are summarized.
Bacteriophage Selection
The first steps in the development of a phage therapeutic are the isolation and selection of safe and efficacious bacteriophages. Phages are found in vast quantities and abundant variety in all environments that harbor bacteria. Phages specific to a certain bacterial strain usually can be isolated from samples containing the host bacteria, such as soil or sewage. Phages are obligate parasites of bacteria. They have been shown to coevolve with their bacterial hosts.(24) Consequently, phages specific to a mutated bacterium have been observed to emerge soon after mutation of the bacterial host.(25)
An effective phage must: (1) successfully bind to the surface of the bacterium; (2) transfer its genetic material into the host bacterium; (3) synthesize progeny virions; and (4) lyse the bacterial cell.
This process of infection is referred to as the “lytic life cycle” as shown in Figure 2. One lytic cycle is completed in about 15–60 min and produces typically 100–200 new phages.(21,26–32) In phage therapy, the concentration of phages in vivo will theoretically grow rapidly if the bacterial host is present in sufficient quantity. When used therapeutically, the high interaction specificity between a phage and the bacterial cell prevents the viral particles from destroying human cells. However, this also limits the usefulness of certain phages due to their extreme selectivity. In selecting a phage, or set of phages for clinical use, the most important criterion is bacterial host specificity. Phage host ranges are limited due to factors such as the inability to bind the host or to transfer DNA, the cleavage of phage DNA by host proteins, or bacterial abortive infection systems.(33) The narrow host range of phages is beneficial, because it avoids the adverse effects on the normal flora observed following broad-spectrum antibiotic treatment.(25) A disadvantage of this specificity, however, is that a panel of phages must be screened on the infecting bacterium prior to administration in order to assess which phages are the most effective in lysing the bacterial cells.(16)
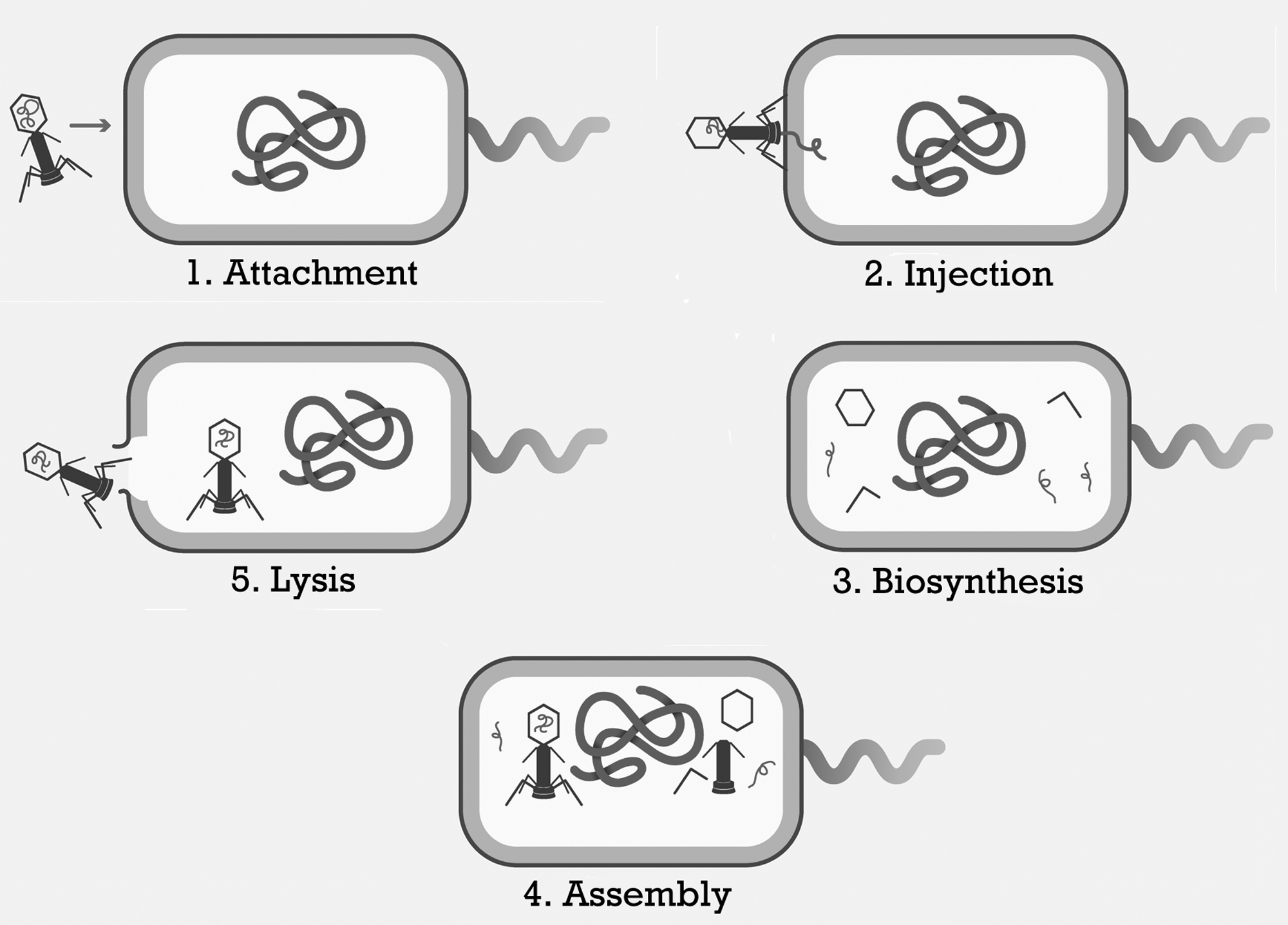
Life cycle of lytic bacteriophages.
To mitigate this issue, phages with expanded bacterial host ranges can be isolated or developed using classical or molecular techniques.(34,35) In addition, multiple phages may be used together in a cocktail formulation, and this is especially useful for polymicrobial infections where multiple bacterial pathogens are present (as might be found in the CF lung).(34) This can be achieved by selecting phages that infect the most common bacterial species or strains associated with a particular infection, or by first determining which bacterial species or strains are involved in the infection and selecting phages for each. A CF patient, for example, might be treated with a cocktail of phages capable of infecting a number of different P. aeruginosa, Burkholderia cenocepacia, and S. aureus strains, because these are common pathogens found in the lungs of CF patients.(36) Phage cocktails can also be used to reduce the chance of bacterial resistance occurring, because phages specific for two different bacterial receptors would require that a single target cell undergo simultaneous mutation of two different receptors, a highly improbable event.
Virulent versus temperate phages
Two other important criteria for phage selection are an obligately lytic lifestyle and the absence of genes that encode virulence factors. Phages can be classified as either obligately lytic (also referred to as virulent) or temperate. Obligately lytic phages follow the lytic life cycle, causing lysis of the bacterial host and release of progeny virions at the end of each infection cycle.(37) In contrast, temperate phages may either lyse the bacterial host and release virions or, alternatively, the phage DNA may be incorporated into the cell as part of the bacterial chromosome or as a plasmid, remaining dormant as a “prophage” through successive bacterial cell divisions until triggered to reenter the lytic life cycle.(37) During lysogeny, the host cell is not killed by the phage and becomes resistant to infection by all other related phages.(38) The integrated prophage genome may occasionally contain sequences that encode virulence proteins such as adhesins or toxins.(39) In the case of Vibrio cholerae (in which virulence factors, cholera toxin, and the toxin-coregulated pilus are found to be prophage-encoded), these genes can have dramatic effects on the pathogenicity of the bacterial host.(40,41) Temperate phages may also transfer bacterial virulence genes via specialized transduction, a process in which host DNA is packaged into progeny virions along with phage DNA following induction.(42)
Genome sequencing and analysis can be used to assess both the lifestyle (obligately lytic or temperate) and the gene content of a phage prior to clinical use. Temperate phages have characteristic sequences that lytic phages lack, including a repressor gene, operator sequences to which the repressor protein binds, and (in most cases) an integrase gene.(38) It should be noted that, in rare cases, phages with these sequences have been identified that do not form stable lysogens (lysogenized bacteria).(43,44) Therefore, assessment of phage lifestyle should include both genomics analysis and screening for lysogens or turbid plaques (characteristic of temperate phages).(42) Virulence genes can also be identified through bioinformatics analysis and may show similarity to proteins that mediate cell adherence, immune evasion, toxin production, or other processes.(39)
The concerns discussed above usually lead to a recommendation to select lytic phages for therapeutic applications. However, several cases have been identified in which phages with temperate lifestyles or virulence genes have been shown to be effective in phage therapy applications. For instance, most phages of Clostridium difficile and the Bcc appear to be temperate.(45,46) Out of necessity, phages used (and shown to be effective) in infection models of these bacterial species are either temperate or putatively temperate.(47–50) Temperate phages have also been shown to be effective against S. aureus, even in a case where the phage carried putative leukocidin and enterotoxin A genes.(51–53) Instead of discarding phages encoding proteins involved in lysogeny or virulence, genetic modification may be an option to improve safety prior to clinical use. Lytic mutants of temperate phages have been shown to be effective against Escherichia coli in a mouse model,(54) S. aureus in human volunteers and a mouse wound model,(55) and B. cenocepacia in an invertebrate model.(48) These phages may be spontaneous mutants (with a characteristic clear plaque morphology) or generated by random or targeted mutagenesis of the operator or repressor sequences.(42,48,55) Although the use of obligately lytic phages prevents lysogenic conversion,(56) virulence genes can be removed from the temperate phage genome such that they do not introduce problematic genetic material upon recombination with the bacterial chromosome during infection.
It has been shown that some phages can penetrate biofilm matrices, or possess surface-exposed enzymes that are capable of degrading the polymeric matrix components of bacterial biofilms.(57,58) Such polysaccharide depolymerases or glycanases are capable of allowing some phages access to bacterial host cells deeper within biofilms, thereby enhancing lysis and degradation of the bacterial biofilm.(59,60) The genes encoding such phage biofilm-degrading enzymes can be transferred to and expressed by other phages in order to enhance phage penetration of biofilms.
In addition to removing lysogeny and virulence genes, or adding genes encoding enzymes capable of enhancing biofilm degradation, other phage components can be genetically manipulated prior to therapeutic use.(61) Such modifications include bacterial host range expansion(35,62) and the introduction of antibacterial toxin genes.(63–65) Engineered phages have been shown to be active against several species in mouse models, including E. coli, S. aureus, and P. aeruginosa.(53,64–66)
Potential host effects
Phages are considered to be biological entities and, when used as therapeutic agents, have the potential to interact with or stimulate the host's immune system. However, phages as antibacterial agents are not unique in this respect. Several approved protein-based pharmaceuticals also have the ability to stimulate an immune response, and chemical antibiotics that achieve their effects through bacterial lysis will cause the release of bacterial cell wall components, including endotoxin. Current dogma suggests that phage particles in an animal or human host are cleared by the reticuloendothelial system within a few days.(54,67,68) However, the immunogenicity of phage particles is likely highly variable, partially depending on where the phages are initially administered (bloodstream versus body surface). For example, Carmody et al. were able to provide a small therapeutic effect to a pulmonary bacterial infection via intraperitoneal phage delivery, suggesting that some phages can easily penetrate different body cavities without activating an anti-phage immune response.(47) Within the lung, phages showed amplification and, at the same time, markers of pathogen-induced inflammation were reduced. Similarly, Merril et al. were able to evolve long-circulating phages that were presumably less immunogenic than parental types, suggesting that some phages may be relatively invisible to an immune response.(54) Although little rigorous information exists about immune stimulation by phages in vivo, the paucity of such data suggests that phage lysis of bacteria in an animal or human host is of little consequence and, at least, does not worsen the host's condition. Several recently completed phage therapy clinical trials suggest that the presence of phages in different body cavities does not enhance the disease state due to immune activation.(16,69) In support of this view, several phage-related products have now passed some regulatory standards and have been classified by the U.S. Food and Drug Administration as GRAS (Generally Regarded As Safe), have been registered with the U.S. Environmental Protection Agency, or have been approved for use by the U.S. Department of Agriculture.(70,71)
Summary
By using conventional screening techniques together with genetic modification, one can select phages for clinical use that are bacterial host-specific, obligately lytic, free of virulence genes, and highly active in vivo. There is little existing data on the human or animal immune response to phages, but also little evidence of phages having a negative effect on disease state.
Bacteriophage Preparation
As discussed, there are already many phages available that are capable of infecting a wide range of antibiotic-resistant bacteria, and more are continually being isolated and characterized. The procedures for growing phages are well described elsewhere; a summary of these preparation steps is provided in Figure 3.
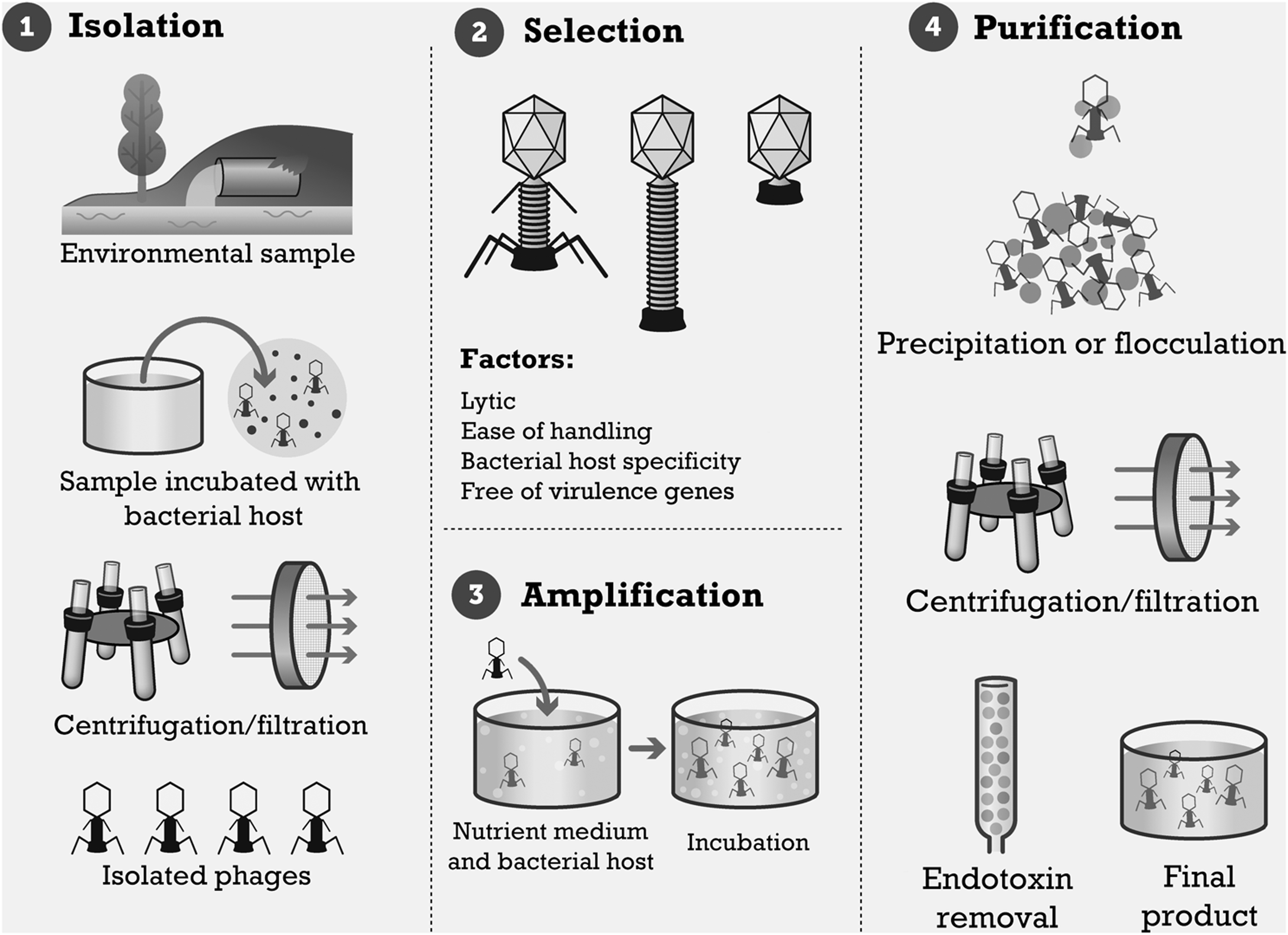
Processing steps in the manufacture of bacteriophage preparations.
Phages are obtained from environmental or biological samples, such as soil, river water, sewage effluent, and fecal matter. One or a panel of bacterial strains is used to propagate any desired phages in the sample before filter sterilization to remove particulate and cellular matter. Samples are often mixed with a culture medium along with the bacterial host, and the entire preparation is incubated to allow phages to bind to potential hosts. If host-specific phages are present in the sample, the bacterial cells will be lysed to release progeny virions. The lysate is then centrifuged or passed through a membrane filter, or both, to elute the phages in a sterilized sample. This sample is then plated against bacterial strains to isolate the single desired phage.(16,72–74)
Once phages have been isolated, they can then be grown to the desired concentration. This is known as phage amplification. As with enrichment, the phage is cultured with the host bacterial strain or strains in a suitable medium and incubated. This can be done in liquid culture or on agar plates, followed by an overlay of sterile water or suspension medium to suspend the phages in solution. Separation of the phage from cellular debris is often carried out by centrifugation and membrane filtration (Fig. 3).
Early phage preparations were often just filtered lysates that still contained bacterial endo- and exotoxins at the point of human delivery.(75) There are no reports of serious side effects due to endotoxins in the early literature covering experimental bacteriophage therapy. Nevertheless, current expectations regarding the safety of biological preparations require removal of bacterial endotoxin from phage preparations before proceeding with any other modifications (i.e., spray-drying, freeze-drying, nebulizing, etc.). Endotoxin is a part of the cell wall of Gram-negative bacteria known as lipopolysaccharide. The lipid component is associated with toxicity, whereas the polysaccharide component is responsible for eliciting an immune response.(76)
Lipopolysaccharide is typically present in initial phage preparations once phages have lysed bacterial host cells. There are a number of commercially available products that remove endotoxin from phage samples. For large-scale preparations, ion-exchange or gel filtration chromatography may be used.(76–78) Another method of removing endotoxin involves a series of phage dialysis and purification steps using an organic solvent such as octyl or butyl alcohol.(79)
Because laboratory-scale methods of bacteriophage growth and purification tend to be labor-intensive and time-consuming, commercial production of phage preparations requires optimization of manufacturing steps during scale-up. The production process for live attenuated virus vaccines has been developed to commercial stage and is now a routine operation.(80) Many of the processing steps used in virus vaccine production are similar to the ones needed for bacteriophage manufacturing.(72) Specific improvements for phage manufacturing processes can be found in the patent literature: Bujanover proposed the use of a semisolid culture medium instead of liquid medium for incubation with the host bacterial strain.(81) This is followed by a repeating process of dilution, centrifugation, and extraction of supernatant to achieve a bacteriophage titer of at least 1010 plaque-forming units (PFU)/mL in the final extract. Lenherr and Bartsch outlined a fully automated purification process.(73) A prepurification step involves passing the unprocessed postamplification phage and bacteria preparation through a rotating membrane prefilter with a low-pressure pump. The filtrate is subjected to subsequent filtration steps with progressively finer pore size. The phages are then washed off the final filter and collected.
Summary
Bacteriophage preparation, from raw environmental samples to purified endotoxin-free phage product, can be accomplished using established biotechnology processes. Similar production processes for viral vaccines have been scaled up and are routinely used in vaccine manufacturing.
Formulation
After the selection of an appropriate phage and preparation of phage stock, the next step in the development of a respirable dosage form is to incorporate the phage into a stable, inhalable formulation.
The reported shelf life of phage preparations varies, and although many phages can be stored for years without any significant loss, not all phages are suited to long-term storage without further stabilization measures. Clark (1962) tested the effect of 2 years of different storage conditions on the preservation of 14 different phages.(82) He found that most phages survived better when stored at 4°C. At this temperature, 10 phages survived best when freeze-dried, four when suspended in liquid, and three when stored in 50% glycerol. More recently, it has been reported that lactococcal phages are stored best at −30°C in a broth supplemented with calcium, but are less stable when freeze-dried.(83) However, Bcc and P. aeruginosa phages KS4-M and ΦKZ, respectively, can be stored without significant titer loss after lyophilizing in a lactose/lactoferrin matrix.(84)
Storage conditions for phage preparations will, therefore, depend on the individual phage, or phages, that they contain. A variety of commercial phage preparations in Georgia are marketed as liquid suspensions with a shelf life of 2 years when stored at 2–15°C(85) and are also available in a lyophilized form with 1.5 years of shelf life at unspecified storage conditions.(86) Given that phages are protein structures, they are potentially susceptible to factors that are known to cause protein denaturation, including temperature, pH, interfacial adsorption, and ionic strength. Table 2 provides a summary of the studies discussed in this section.
Temperature
It is well known that the application of high temperatures leads to thermal denaturation of proteins, a fact that can be used for sterilization or pasteurization purposes. Phages show typical behavior in response to short-term temperature exposure: S. aureus-specific phages FH5 and FA72 were found to maintain initial titer (>4×105 PFU/mL) after suspension in ultra-high-temperature–processed, whole-fat milk when stored at 4°C for 8 hr, showed approximately 20–25% titer loss at 37°C, and showed complete loss at 72°C.(87) Caldeira and Peabody demonstrated thermal stability of E. coli-specific MS2 phages in storage media, from 20 to 60°C, after 2-min exposure to temperature.(88) Briers et al. identified the peptidoglycan hydrolase gp181 as an important part of phage ΦKZ infectivity against P. aeruginosa, and found that its activity gradually declined from 100% to 0% when exposed to temperatures of 25–90°C for 60 min.(89) The study of phage survival is well established in the dairy industry, where the presence of phages can inactivate the bacterial cultures required for fermentation. For example, the Lactococcus lactis phage P008 experienced 6 log10 titer loss when held at 72°C for 30 min(90); the heat-resistant variant phage P680 experienced a 2 to 3 log10 titer loss when held at 90°C for 10 min.(91) Short-term temperature exposure is not a concern in the manufacturing of phage preparations, because high-temperature exposure is unnecessary or avoidable in all processing steps. However, temperature excursions during transport or administration may need to be considered.
pH
The pH is an important formulation parameter that is commonly controlled in feedstock preparation for further processing steps such as freeze-drying. In general, phages are well adapted to pH ranges typically encountered in living systems. Four P. aeruginosa-specific phages were found to achieve 100% survival at pH 7 and 80–100% survival at pH 9, but declined in phage activity when pH was decreased to 1.5.(92) However, formulation pH may affect the charge carried on protein surfaces, resulting in an alteration to electrostatic and hydrophobic interactions, and a change in protein conformation.(93) For MS2 bacteriophages, pH conditions close to or below the isoelectric point were shown to favor phage aggregation, associated with a greater than 1 log10 loss in PFU count.(94)
Ionic strength
The ionic strength of a formulation may affect the osmotic pressure on bacteriophages in a suspension; if the difference in osmotic pressure inside and outside the phage is too great, the phages may be destroyed by osmotic shock.(95) This may be of particular concern if the ionic strength changes rapidly, for example, during removal of water in drying processes. The peptidoglycan hydrolase gp181 used in phage ΦKZ infectivity maintained optimal activity at a pH of 6.2 and ionic strength of 140 mM; variation in pH and ionic strength in either direction (pH 5–8, 20–320 mM) only produced a significant decline in enzymatic activity.(89) Golshahi et al. observed a 74% drop in KS4-M bacteriophage titer when prepared in a hypotonic suspension (105 mOsm), compared against an isotonic (282–290 mOsm) suspension.(96)
Interfacial adsorption
When presented with an interface, e.g., during filtration or in the process of dry particle formation, proteins may unfold to assume the conformation of lowest energy; its hydrophobic regions are exposed to the hydrophobic phase (air or organic solvent) and its hydrophilic regions (charged and polar groups) to the hydrophilic phase (aqueous or polar solvent). This has ramifications for the design of a stable bacteriophage formulation, where the phages may need to be protected from the liquid–air interface. Pharmaceutical formulations often use nonionic surfactants to prevent interfacial adsorption of proteins.(97)
In addition to thermal and chemical stresses, phages may be susceptible to damage by mechanical stresses induced during processing. In particular, shear stress from high-speed mixing, filtration, and centrifugation is a potential source of denaturation.(98) Short-term and long-term stress on bacteriophages during processing and storage apply to phages regardless of their mode of delivery, and standard techniques for stabilization such as cryoprotection and desiccoprotection may be applied. For respirable dosage forms, additional criteria must be met. The final dosage form must be suitable for efficient inhalation and delivery to the appropriate disease target.
Summary
Formulation of stable phage dosage forms appears feasible, given that over-the-counter phage preparations are already available. Formulation variables to consider include pH, temperature, osmotic pressure, ionic strength, and mechanical stress. However, formulation of biological actives into a dosage form suitable for respirable delivery remains an active area of research.
Phage Delivery
The efficacy of phage delivery to an infected respiratory tract is reliant upon a number of factors common to respirable dosage forms. Two significant factors are their aerodynamic behavior under inhalational airflow and the intended target for delivery.
Aerodynamic particle characteristics
Aerodynamic particle diameter is often used in in vitro aerosol studies to estimate regional deposition in the respiratory tract. The actual deposition pattern of particles in the airways is obviously also affected by device and patient factors, such as the breathing profile and individual airway geometry. As depicted in Figure 4, for typical breathing patterns and normal airways, it is generally accepted that particles with an aerodynamic diameter between 1 and 5 μm are respirable, whereas most particles larger than 5 μm are expected to deposit in the extrathoracic airways, and particles smaller than 1 μm (but larger than 100 nm) are liable to be lost in exhalation.(99) From aerodynamic particle diameter measurements, the parameters of fine particle dose (mass of delivered particles smaller than 5 μm), mass median aerodynamic diameter (MMAD; diameter at which 50% of the delivered mass is smaller than this value), and geometric standard distribution can be used along with delivered dose to predict the amount of aerosol delivered into the lung and the delivery efficiency.(100)
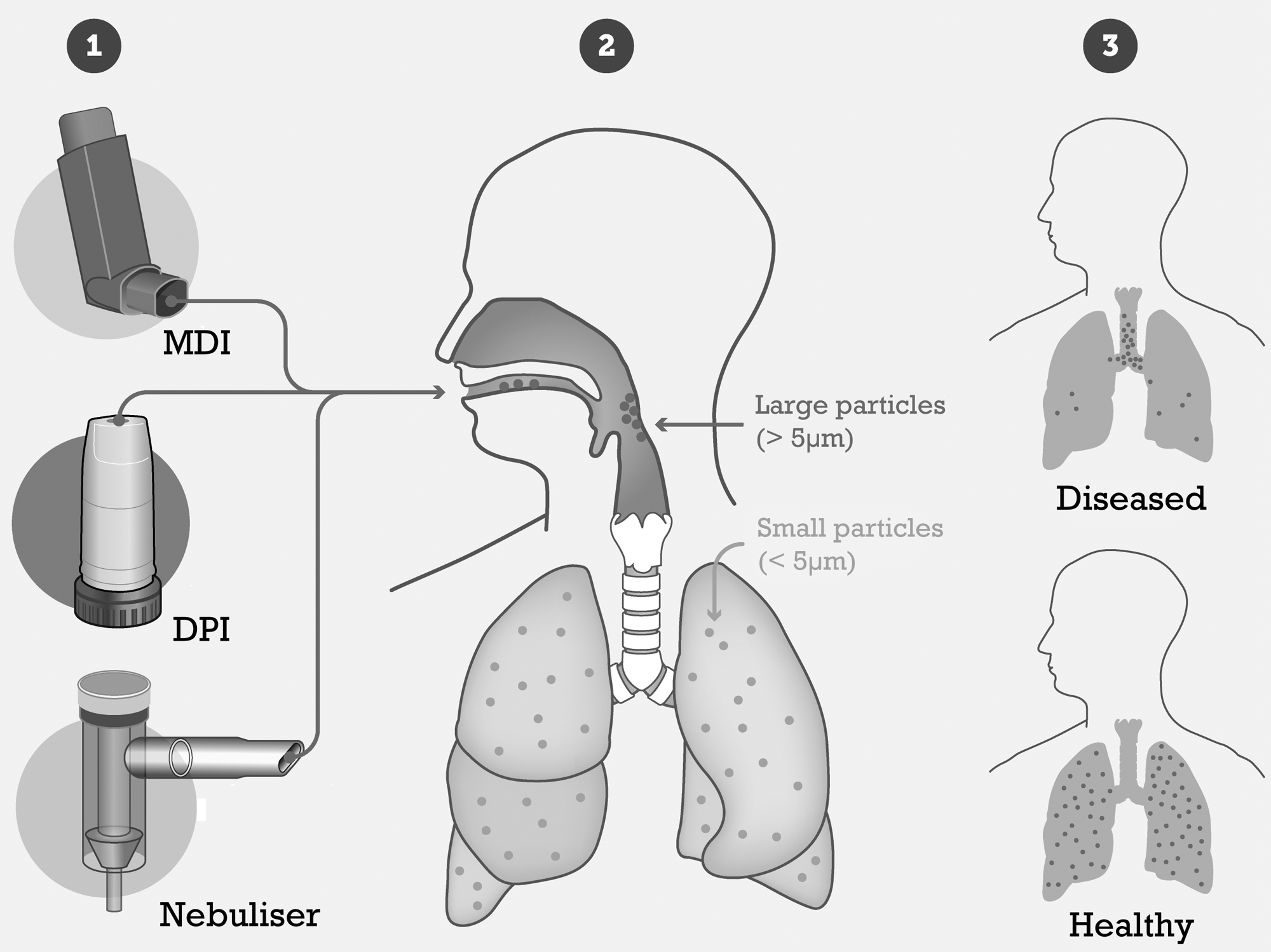
Common delivery device options [metered dose inhaler (MDI), dry powder inhaler (DPI), or nebuliser], and delivery targets in healthy and diseased lungs.
Delivery target
While in vitro studies of aerosol particle size distribution often attempt to correlate fine particle dose to lung deposition, the architecture and airways surface liquid composition of an acute or chronically diseased lung weakens this in vitro–in vivo correlation further. If we were to take CF as an example, features of the disease, such as bronchiectasis, peribronchial thickening, mucus plugging, and air-trapping,(101) would be expected to alter the aerodynamic profile of the pulmonary system, as shown in Figure 4, not only due to morphological changes, but also as a result of changes in inhalation profile.
The results of chest radiography studies in CF patients show a more complicated range of disease presentation, making it difficult to mathematically model the diseased respiratory system.(100) Kaza et al. identified diffuse bilateral disease from chest radiographs in 62% of the adult patients studied (n=68), compared with 28% with predominant upper lobe disease and 7% with lower lobe disease.(102) In comparison, a small study of 17 children younger than 4 years of age found six patients with the greatest disease in the right upper lobe and another six in the right lower lobe, but the left upper lobe was also qualitatively categorized as having the least amount of disease in six subjects.(103) Another study of 60 children (aged 6–10 years of age) found bronchiectasis and mucous plugging to be distributed similarly between the left and right, and upper and lower, lobes.(104)
Nevertheless, numerical models have been devised to assist in vitro prediction of aerosol deposition in CF patients. In one example, deposition of coarse particles (>5 μm MMAD) by impaction was predicted to be two to three times higher in the large apical and basal airways (generations 1 through 12) of CF patients than in those of healthy subjects.(105) However, to keep the model workable, assumptions were made in their simulation: disease was assumed to progress from the small to large airways, sedimentation and impaction were the only deposition mechanisms included, and the disease was more severe at the apex.
Based on other aerosol formulations in CF, and due to uncertainties in predicting deposition patterns in diseased lungs and considerable patient variability (both intersubject and temporal intrasubject variability), it seems advisable to design bacteriophage dosage forms with an MMAD smaller than approximately 5 μm for lung infections, and an MMAD larger than approximately 5 μm for infections of the extrathoracic airways.
At the time of this publication, three formulation techniques have been used to manufacture a respirable phage dosage form: suspension of phages in a liquid medium, freeze-drying, and spray-drying. These formulations have then been loaded into inhalation devices, intended for delivery into the human respiratory tract. This section will outline the phage formulations that have been successfully produced and the outcomes of the in vitro aerosol testing performed for these formulations (see Table 3 for a summary of the studies discussed in this section).
Suspension formulations
So far, the production of liquid suspension for direct phage delivery into the respiratory tract has been for nebulization. To the best of our knowledge, no work has been published reporting suspensions of bacteriophage in propellants for metered dose inhaler delivery. Animal studies and experimental clinical work have adopted nasal and pleural instillation and insufflation of phage suspensions as a method of delivery.(47,106,107)
Commercial nebulizers for inhalational drug delivery employ different types of aerosol dispersion,(108,109) including jet (air-blast) nebulization, vibrating mesh plate nebulization, ultrasonic nebulization, as well as colliding jets. With jet nebulization, compressed air is passed through a gap in a thin tube, leading to a Venturi effect that creates a pressure gradient that draws the nebulizer solution or suspension up a feed tube. Droplets are generated via a viscosity-induced instability,(100) whereby nonlinear growth of surface waves and subsequent drop breakup and filtering by impaction on baffles occur. Vibrating mesh nebulization may be carried out by two methods: (1) a piezoelectric crystal vibrates at high frequency, transferring the vibration to a transducer horn that pushes the liquid through a mesh plate (“passive”); or (2) a piezoelectric element directly vibrates a mesh plate, causing micrometer-range vertical displacement of the mesh that generates droplets from the liquid drawn through (“active”).(110,111) Ultrasonic nebulization does away with the mesh plate altogether, and involves the oscillation of a piezoelectric crystal transducer, which transfers the oscillation energy to the liquid. Droplets are formed by the breakup of surface capillaries or the collapse of cavitation bubbles within the liquid.(109,111)
Nebulization was the mode of delivery in the early reports on phage inhalation reviewed in the introduction.(19,20) However, few device and formulation details are given, and information on possible titer loss during nebulization cannot be derived from these publications. Phages have been nebulized to test the efficacy of biological respirator filters, alongside aerosolized bacteria, fungi, and viruses (e.g., Balazy et al.,(112) Eninger et al.(113)). However, the focus of these studies is typically on particles in the most penetrating size range (a few hundred nanometers in diameter) and normally involves evaporated nebulized aerosols usually produced by the standard Collison nebulizer, which is commonly used in laboratory studies in occupational hygiene. However, such submicrometer particle sizes are not optimal for respiratory delivery due to their low efficiency of deposition. In addition, the Collison nebulizer does not match current commercial nebulizer designs in its ability to deliver therapeutic aerosols to the human respiratory tract. For example, Liu et al. used the six-jet Collison nebulizer to aerosolize D29 phages (for M. tuberculosis).(114) With an initial phage titer of 108 PFU/mL, the phage suspension was aerosolized at 10 L/min, into a chamber for 10 min, before the aerosol was sampled at 12.5 L/min for 5 min in a liquid impinger. Regardless of the suspension medium (PBS or saline), the sampling medium (PBS, saline, or deionized water), or relative humidity in the chamber (25, 55, or 85%), there was a 2 to 3 log10 titer loss upon collection of the aerosolized phage, supporting the conclusion that the Collison nebulizer may not be the best choice for phage lung delivery.(114)
A study by Golshahi et al. reported on nebulizer suspensions designed with the intent to deliver phages to the respiratory tract.(96) The phage selected for the study was KS4-M, which is selective for one strain of the highly antibiotic-resistant Bcc. KS4-M was prepared to an initial phage titer of 2.15±1.63×108 PFU/mL. Two commercial nebulizers were used in this study: the Pari LC Star jet nebulizer, and the Pari eFlow vibrating mesh nebulizer.(96) The eFlow system is of particular interest, given that it is has been suggested that its active vibration nebulization subjects the liquid to less shear stress,(115) a feature that could potentially reduce phage titer loss from nebulization. Each nebulizer was loaded with 2.5 mL of the KS4-M suspension and aerosolized under a simulated inhalation profile generated by a breathing machine (tidal volume=800 mL, respiratory rate=14 breaths/min, duty cycle=0.5). No significant difference in total nebulizer output was found between the eFlow and LC Star systems, with both producing about 108 PFU. This equates to less than 1 log10 titer loss, which is an ideal outcome for a respiratory delivery method; although the vibration mesh nebulizer did not appear to give an advantage in phage survival, both nebulizers demonstrate a clear advantage over the Collison nebulizer used by Eninger et al.(113) There were also no significant differences between the two nebulizers for the KS4-M droplet size distributions, with a mass mean geometric diameter of 5–6 μm.
The distribution of these nebulized droplets in the lung was predicted by the incorporation of particle size distribution and breathing pattern data into a numerical respiratory tract deposition model. Alveolar deposition was approximately 3×107 PFU for both systems. These distribution data were then used in a follow-on model of airway surface liquid in the lungs to estimate KS4-M concentration within the different lung generations, dependent on mucus production and velocity. For both the LC Star and eFlow nebulizers, concentration decreased with conducting airway generation, from 1.9–2.2×107 PFU/mL (generation 1) to 6.0–7×106 PFU/mL (generation 13). It should be noted that although these deposition predictions are promising, the numerical models are based on healthy patient lung architecture.
Dry powder formulations
In designing an inhalable dry powder formulation, an important factor for consideration is the ability of the delivery device to entrain and deaggregate the powder, which requires a dispersible powder. Most dry powder inhalers (DPIs) are of the passive type, requiring the patient's inhalation effort to generate a sufficient air flow rate to disperse the dry powder into the respiratory tract.
Although milling is commonly used to produce powders for inhaled aerosol formulations, destructive forces in mills may result in reduced survival of phage formulations.(84,116,117) Instead, lyophilization and spray-drying have been used to produce dry powder formulations intended for inhalation therapy. To the best of our knowledge, only two published articles have extended bacteriophage dry powder formulation studies as far as in vitro aerosol testing: one using a lyophilized dosage form and the other a spray-dried powder.
Lyophilization (freeze-drying)
Lyophilization has long been used for the preservation of commercial protein formulations, including vaccines, antihemophilic factors, human growth hormone, and interferons.(118) Lyophilization has also been used to preserve phages; however, phages exhibit different stabilities (short- and long-term) depending on the formulation contents and method of lyophilization,(119–121) and there are few data specifically dealing with lyophilization of phages infecting problematic respiratory bacterial species such as P. aeruginosa and the BCC.
Traditional lyophilization involves three stages: freezing, primary drying, and secondary drying. During the freezing stage, the solvent crystallizes amongst solid amorphous or crystalline solute. Solid solvent sublimation occurs during primary drying, and moisture desorption during secondary drying, leaving a solid porous “cake” of the solute.(122) Excipients such as polyethylene glycol (PEG), trehalose, mannitol, and sucrose are commonly added as cryoprotectants, protein stabilizers, and bulking agents, to give the cake pharmaceutically acceptable physical properties.(122,123) Related to traditional lyophilization, spray freeze-drying, whereby primary freezing occurs upon spraying into cold liquid, has been proposed.(124) In addition, atmospheric spray freeze-drying, where primary freezing occurs upon spraying into cold gas in order to ease commercial scale-up issues, has been used to prepare aerosol powders of bacteria(125) with 90% survival rates in the prepared aerosol powder.
Using traditional lyophilization, Golshahi et al. carried out a study on freeze-dried phage formulations designed for respiratory delivery.(84) The phages KS4-M (for a Bcc strain) and ΦKZ (for P. aeruginosa) were each formulated with lactose and lactoferrin. Lactose served as a carrier and bulking agent.(84) Lactoferrin, a protein abundant in airway surface liquid, has been shown to inhibit P. aeruginosa biofilm formation.(126,127) In addition, lactose and lactoferrin were selected as cryoprotectants and stabilizers for the phages. A 60:40 % w/w ratio of lactose to lactoferrin was selected on the basis of its optimal aerodynamic particle size distribution and protection in preliminary studies. Viability tests post lyophilization demonstrated a 2 log10 titer loss for KS4-M and a 1 log10 loss for ΦKZ. The authors also reported no further phage loss after a milling procedure in a mixer mill (without the use of beads) to deagglomerate the lyophilized powders.
The lyophilized powders were also subjected to in vitro aerosol testing; 10 mg of powder was dispersed from an Aerolizer DPI into the Alberta idealized throat and filter at a vacuum air flow rate of 60 L/min, where the mass of powder recovered from the filter was representative of respirable dose. The results showed an acceptable titer drop from capsule dose to respirable dose, with the maximum loss observed being 1.2 log10 for KS4-M, and 0.84 log10 for ΦKZ. Respirable doses of 3.4±2.5×106 for KS4-M and 1.9±0.6×107 for ΦKZ were achieved.
The phage powders were stored at 21±2% relative humidity and temperatures of 4°C and 21°C for 3 months as part of a short stability study. There was no evidence of titer loss for either KS4-M or ΦKZ, indicating that phage activity was preserved. Long-term stability results were not reported.
Alfadhel et al. did perform a 12-month phage stability study on a formulation designed for nasal delivery.(128) Included in the formulation besides an S. aureus-specific phage was hydroxypropylmethylcellulose (HPMC), to alter viscosity and increase residence time in the nasal cavity, and mannitol, as a lyoprotectant and bulking agent. Active phage titer was decreased by 1 log10 after lyophilization. During storage of the freeze-dried powder at 4°C, there was a further 1 log10 loss after 1 month, another 1 log10 loss after 2 months, and a total of 3 log10 loss from the beginning of refrigeration to the end of the 12-month period.(128) Ultimately, the acceptability of this magnitude of titer loss depends on its variability, the necessary delivered dosage, and whether a formulation with sufficiently high initial titer can be practicably prepared to offset the loss.
Furthermore, the addition of mannitol did not appear to affect phage survival, with the formulations containing HPMC (1 or 2% w/v) as the only excipient performing similarly to those with both HPMC and mannitol. A mannitol-only phage formulation was found to exhibit 6 log10 titer loss just after lyophilization. These results pointed toward HPMC acting to maintain protein conformation, and the authors suggested the stability of amorphous HPMC to be crucial (the glass transition temperature for lyophilized HPMC was approximately 187°C).(128) Although the formulations tested in this study were intended for nasal delivery, they demonstrate that high-molecular-weight polymers may have a future role in maintaining phage activity during lyophilization and storage.
Spray-drying
Spray-drying is an established technique for the manufacture of pharmaceutical products.(129) As shown in Figure 5, a liquid feedstock is atomized, typically using atomization gas in a twin-fluid nozzle, and mixed with drying gas in a drying chamber. The atomized droplets rapidly evaporate, and dry particles with a respirable particle diameter are produced, separated from the gas stream, e.g., in a cyclone, and collected.

Schematic of a spray-dryer for respirable powders.
When drying biological material, the design of the spray-drying process(130,131) must be conducted in a way that minimizes stresses, including shear, temperature, and desiccation stress.(132) Low-temperature spray-drying processes for heat-sensitive biological pharmaceutical ingredients have been developed.(133–135)
Walbeck submitted a patent application detailing the use of a rotary atomization spray-dryer system for the production of E. coli O157:H7-specific phages in dry powder.(136) The process used an inlet temperature of 200–220°C and an outlet temperature of 90–130°C. The authors predicted the powders to contain 2–4×109 PFU/mL; the actual phage titer of the spray-dried powders was 1.1–1.8×102 PFU/mL, a 7 log10 titer loss.(136) Given that existing literature has established that some phages are thermally labile at temperatures below 100°C (as described above), it is unsurprising that such high outlet temperatures would result in significant titer losses.
Matinkhoo et al. presented a study on spray-dried bacteriophage powder formulated for inhalation from a DPI.(137) The authors selected the Bcc phage KS4-M and the P. aeruginosa phage cocktail ΦKZ/D3 for study. Each phage or phage cocktail was formulated with trehalose for protection against thermal stress and desiccation, and leucine as a dispersibility enhancer in the ratio 76:19, with other excipients in the remaining 5%. Given the considerations to be made about phage inactivation from thermal stress, a relatively low spray-dryer outlet temperature of 40–45°C was selected. A vibrating mesh atomizer with 4μm mesh orifice diameter was used to produce 7μm droplets.(137)
The authors reported less than 1 log titer loss after spray-drying for all tested phages, regardless of formulation composition.(137) The ΦKZ/D3 cocktail was particularly resilient, with less than 0.5 log10 titer loss in all cases. These results not only indicate that inactivation by thermal stress was largely prevented, but also that the other factors for inactivation, like shear stress or desiccation stress, were not significant influences for the chosen formulations. Also reported was a less than 0.15 log10 titer loss for ΦKZ/D3 spray-dried powder after 3 months of refrigerated storage. However, as with the freeze-drying studies, long-term stability results were not reported, but would be necessary to advance the phage formulations into clinical trials.(137)
Matinkhoo et al. also subjected the spray-dried powders to in vitro aerosol testing, using an Aerolizer DPI and the same methodology as described for the lyophilized powder produced by Golshahi et al.,(84) but with 25–28 mg of powder loaded into the capsule.(137) The respirable dose was ∼70% of loaded powder mass, regardless of phage type. Phage titers for the respirable dose were ∼107 PFU for ΦKZ/D3, and ∼108 PFU for KS4-M. Separate particle size measurements with the Andersen Cascade Impactor showed the KS4-M and ΦKZ/D3 in leucine-trehalose-casein formulations to have an MMAD of <3 μm, suggesting that the powder is highly dispersible and capable of reaching the smaller airways.(137) An efficacious dose for local phage treatment of lung infections has not been established, but the titers reported in this study are two to three orders of magnitude higher than the minimum titer in commercial phage preparations for P. aeruginosa infections distributed in Georgia.(85)
Summary
The feasibility of efficiently delivering phages to the lung has been demonstrated using different processing and device options. More work in developing a stable respirable dosage form is needed, and long-term stability of such dosage forms still needs to be evaluated.
Reports on Safety and Efficacy in Animals
The study of aerosol phage therapy in animals has been a relatively recent addition to the body of research being conducted in phage therapy, and further studies need to be conducted. The aerosol phage therapy animal studies reviewed here (Carmody et al.,(47) Debarbieux et al.,(138) Morello et al.,(106) Alemayehu et al.(59)) all follow a similar protocol. Mice are infected and treated via an intranasal instillation of the liquid. Intranasal inhalation requires the liquid containing either the bacteria or phage to be dripped onto the nares of the mouse, and from there the droplets of the liquid are inhaled into the lungs.
Although intranasal instillation is a much easier delivery route to use in animal studies because there are no additional aerosol generators or delivery systems required, it should be noted that it does not accurately mimic aerosol drug delivery in clinical environments, and as such it is difficult to extrapolate the results of these studies to a prediction of aerosol therapy efficacy. Indeed, a previous study has shown that intranasal instillation of bacteria does not mimic aerosol particle deposition within the lungs of mice: Halperin et al. demonstrated that mice given Bordetella pertussis respiratory infections via two methods, either intranasal inhalation or a whole-body exposure chamber, had very different bacterial lung counts.(139) The mice receiving intranasal instillation showed a far higher degree of variability in bacterial load in comparison with those receiving aerosolized bacteria (a 1,000-fold variability as compared with a five-fold variability, respectively). The mice receiving intranasal instillation also showed a far greater variability in bacterial distribution within the lungs (right lung, 43–84%; left lung, 16–57%) in comparison with those receiving aerosols via the whole-body exposure chamber (right lung, 60–68%; left lung, 32–40%).(139) This may also translate into higher variability when treating mice in phage therapy studies, and it would be of interest to see additional studies performed using a whole-body exposure chamber or nose-only inhalation chamber, in order to observe the effect of aerosol phage delivery method on treatment efficacy.
Debarbieux et al. demonstrated the efficacy of aerosol phage therapy via intranasal instillation for treating acute respiratory infections using a bioluminescent strain of P. aeruginosa PAK.(138) Provided that the phage titer was high enough and treatment was given soon enough, the phage treatment proved effective. Untreated mice all died within 48 hr of infection, whereas those treated 2 hr after infection with a multiplicity of infection (MOI) of 10 all survived to the end of the trial (12 days). Twenty-four hours after treatment, the bacterial load in treated mice was already 6 log10 lower than in untreated mice; 48 hr after treatment, the inflammation levels in the phage-treated mice (as indicated by tumor necrosis factor-α and interleukin-6) had decreased to baseline levels, whereas levels remained high in untreated mice.(138)
Morello et al. tested the efficacy of phage therapy via intranasal instillation in treating a P. aeruginosa strain isolated from a CF patient.(106) Mice were treated 2 hr after infection with a high enough phage titer to provide a 95% survival rate (MOI 100). These mice survived to the end of the 16-day trial as opposed to untreated mice, which had 0% survival by 2 days post infection. In addition to survival and an approximately 3 log10 reduction in bacterial lung titer 20 hr after infection, treated mice also had significantly reduced inflammation and significantly reduced lung damage (as demonstrated through histology). Interestingly, immunohistochemistry performed 20 hr after infection revealed that untreated mice had bacteria distributed through multiple areas of the lung, including the macrophages, extracellular space, and alveoli, whereas treated mice were found to have few bacteria remaining in the lungs, and these were mainly localized in lung macrophages.(106) This demonstrates that the immune system works in conjunction with the action of phages to remove bacteria from the lungs.
Debarbieux et al. and Morello et al. both demonstrated that phages could protect mice from P. aeruginosa infection. All mice receiving phage either 1 day (Debarbieux et al.) or 4 days (Morello et al.) prior to infection were protected from death.(106,138) Although this effect may not be as clinically significant as infection treatment, it demonstrates the efficacy of phage usage to prevent infection.
Additionally, Alemayehu et al. demonstrated that a phage cocktail of ΦNH-4 and ΦMR299-2 delivered 2 hr after infection via intranasal instillation was able to clear P. aeruginosa respiratory infections in mice.(59) Two separate bioluminescent strains of P. aeruginosa (MR299 and NH57388A) were delivered intranasally, and the bioluminescence within the lungs was measured at 2-hr intervals up to 8 hr after infection. The luminescence within the lungs (which is proportional to bacterial concentration) showed a significant decrease in phage-treated mice, whereas untreated mice showed a three-fold increase in luminescence intensity.(59)
Carmody et al. studied phage treatment via intranasal instillation in a respiratory infection of B. cenocepacia AU0728.(47) Phages were administered 24 hr after infection via either an intranasal or intraperitoneal route. The bacterial load was determined within the lungs of all mouse groups 48 hr after treatment. In contrast to the three studies reviewed above (Debarbieux et al.,(138) Morello et al.,(106) Alemayehu et al.,(59)), phage delivered intranasally did not produce a significant reduction in bacterial load within the lungs (approximately 1 log10 reduction), whereas phage delivered intraperitoneally did significantly reduce the bacterial load (approximately a 2 log10 reduction), even though a higher phage titer was isolated from mice treated intranasally. The authors suggest that these results indicate that phage therapy in lung infections is more effective when administered systemically rather than topically.(47) There are several reasons why these results may have been obtained. With respect to the reference described above (Halperin et al.(139)), intranasal phage application does not give phage aerosol deposition in the lungs. As we have observed the loss of aerosolized phages out of the lung into the peritoneum (unpublished data), it is not inconceivable that phages can cross from the peritoneum into the lungs. In this case, it appears that peritoneal injection of high titers of phages was more effective than the ineffective intranasal deposition of phages on the mouse nares.
In either case, the 1 to 2 log10 drop in bacterial numbers observed by Carmody et al.(47) may not be an overly effective treatment if the bacterial load is high, and the load is not reduced far enough for the human immune system to deal with the remainder of the infection. One could argue that, in either condition, phage therapy in this study was not efficacious. It is also interesting that more than one phage was not tested, as we have observed notable differences in phage activity in vivo, as compared with their in vitro activity. Finally, due to the controversial nature of these findings, it is unfortunate that these experiments were not repeated with other bacterial species (such as P. aeruginosa) in order to determine if intraperitoneal phage administration would have a similar effect with a proven and published phage therapy candidate.(23,138)
Summary
Animal studies of phage therapy have been limited to intranasal delivery. Although these studies have demonstrated varying levels of efficacy in reducing bacterial load and increasing survival, work is still needed to investigate the safety and efficacy of phage aerosol delivery into the lungs.
Reports on Safety and Efficacy in Humans
Although there have been studies investigating aerosol phage formulations (see Suspension formulations and Dry powder formulations), there are relatively few reports of aerosol phage therapy in humans. However, case studies have shown promising results (see Table 1). In Eastern Europe, it is often applied to infections with antibiotic-resistant strains, and typically 80–90% of cases are cured. This includes phage treatment for respiratory tract infections.(23) Ślopek et al. reported treatment success between 82 and 92% for different patient groups with respiratory infections treated with oral or locally administered phages.(107) Successful treatment of a pediatric CF patient was also reported recently by Kvachadze et al.(140) In this case, phage cocktails were administered to the lung via a nebulizer. However, no controlled clinical trials have been performed to evaluate aerosol phage delivery to patients. Evidence for the clinical safety of phage therapy in humans may be found in studies on oral and topical phage treatment. Four recent trials will be reviewed (Bruttin and Brüssow,(141) Merabishvili et al.,(16) Rhoads et al.,(142) Wright et al.(69)). One of these trials (Wright et al.) also addresses phage therapy efficacy.
The first of the recent phage administration human safety trials in the English literature was performed by Bruttin and Brüssow (2005).(141) Fifteen volunteers were given either a placebo, a low-phage titer preparation (103 PFU/mL), or a high-phage titer preparation (105 PFU/mL) orally on 2 consecutive days followed by 5 days without phage administration. This was repeated for the following 2 weeks with each patient receiving a different phage preparation or placebo each week. Throughout the study, five adverse effects were reported; however, there was no difference in the number of adverse effects in the placebo, low-phage titer, and high-phage titer groups, and the effects were deemed to be unrelated to the study. Phages were never recovered from stool samples prior to phage administration or when the placebo was administered. However, phages were recovered from stool samples in a dose-dependent manner after phage administration, demonstrating that the phages were able to survive transit through the gastrointestinal system. Interestingly, the presence of phage T4 did not correlate with a decrease in natural E. coli counts in the stool samples, suggesting that the phage did not affect the natural microflora of the volunteers, albeit as measured by numbers of only one bacterial species. Throughout the study, there was no change in serum alanine aminotransferase and aspartate aminotransferase levels, which are liver enzymes that would have indicated an increase in liver toxicity. There was also no detected immune response to the phages, demonstrating the safety of phage administration.(141)
A phase I safety trial was performed by Rhoads et al. to ensure that the application of WPP-201, a phage cocktail containing eight lytic phages targeting S. aureus, P. aeruginosa, and E. coli, would not pose any health risks to patients with venous leg ulcers.(142) Thirty-nine patients completed the 24-week trial, with every patient receiving the same compression therapy and wound dressings, with the exception that approximately half received WPP-201 applied topically, whereas the controls received only a sterile saline application. Phage application occurred weekly for 12 weeks, while dressings were changed three times weekly for the same duration. Patients were monitored weekly for the first 12 weeks of the study, and data collected included wound size and severity, vital signs, and adverse events, which may or may not have been related to the phage application (adverse events included a variety of infections, injuries, medical procedures, pain, and cardiovascular issues). Wound bacterial counts, blood chemistry, and blood cell counts were performed every other week as well. Two additional assessments were made on weeks 16 and 24. There were no statistically significant differences between the test and control groups in either the number of adverse events experienced or the frequency with which wounds healed. This phase I clinical study did not set out to evaluate phage efficacy, but instead appraised the safety of phage application. With this objective successfully completed, the researchers can now proceed to phase II studies, which will evaluate the efficacy of WPP-201.(142)
Merabishvili et al. developed and began evaluating BFC-1, a phage cocktail consisting of three phages that targeted P. aeruginosa and S. aureus strains isolated from burn wounds of patients in the Burn Centre of the Queen Astrid Military Hospital in Belgium.(16) The cocktail was sprayed onto the burn wounds of eight patients using a micromister. None of these patients was reported to experience any adverse events resulting from the phage application.(16)
Chronic otitis caused by P. aeruginosa can be problematic to treat, as P. aeruginosa is highly antibiotic-resistant. Wright et al. conducted phase I/II trials to evaluate the treatment of this infection using Biophage-PA, a cocktail of six phages specific for P. aeruginosa.(169) The 24 volunteers in this study, each with a history of chronic otitis lasting 2 to 58 years, had P. aeruginosa infections sensitive to Biophage-PA prior to participation. A single dose of either Biophage-PA or placebo was applied directly into the ear of each participant. The median bacterial counts significantly decreased in phage-treated volunteers on days 21 and 42 (swabs were taken on days 0, 7, 21, and 42), whereas there was no significant decrease in placebo-treated volunteers. This study demonstrated the advantage of administering a self-replicating treatment, as the mean phage count isolated on days 7, 21, and 42 was 1.27×108 PFU, whereas the initial phage count administered was 6×105 PFU. The phage persisted at the site of the infection for an average of 23.1 days, and the phage was cleared in cases where the infection was successfully treated. Each case was also scored on a visual analogue scale (VAS) by the patient and the physician. Patients scored the infection on discomfort, itchiness, wetness, and odor, whereas the physician scored the infection on inflammation, ulceration/granulation/polyps, discharge quantity, discharge type, and odor. When the patient VAS scores from days 7, 21, and 42 were averaged, there was a significant decrease from days 0 for discomfort, itchiness, and wetness, as well as overall patient VAS scores, whereas placebo-treated volunteers did not show any significant reductions. When the physician VAS scores from days 7, 21, and 42 were averaged, there was a significant decrease in inflammation, ulceration/granulation/polyps, discharge type, and odor, as well as overall physician VAS scores, whereas the placebo group did not show any significant reductions. As with the other clinical trials reviewed, none of the adverse events reported during the course of the trial was deemed to be related to phage treatment. This study provided sufficient evidence of phage therapy efficacy to warrant the design of a phase III clinical trial.(69)
It is interesting to note that these studies delivered a range of phage titers to the infected sites and, in the phase I/II trial, only one phage dose was required for treatment.(69) This highlights a unique difference in the pharmacokinetics of phage therapy in contrast with traditional chemical antibiotics. Traditional antibiotics must be administered at regular intervals as they are metabolized and excreted. In some situations, similar delivery schedules for phages are used. This is known as passive phage therapy, because the treatment does not rely on phage replication within the host bacterium.(143) In some situations, a single treatment is sufficient because phages can replicate at the site of infection, allowing for the continued release of new antibacterial progeny. This is known as active phage therapy.
The bacterial density at the site of infection may also affect the treatment strategy. Active phage therapy is effective in areas of high bacterial density, whereas passive phage therapy may be effective in areas of reduced bacterial density.(30) Interestingly, mathematical modeling has demonstrated that the timing of phage administration is also very important.(143) As expected, delayed administration of phages may be detrimental to treatment; however, treating an infection too early (e.g., low bacterial numbers) may also be detrimental. This, again, relates to the density of bacterial hosts available at the site of an infection. A very low density bacterial load may be more treatable once it has replicated to the point at which the density is high enough for effective active phage therapy. The studies reviewed above (Bruttin and Brüssow,(141) Merabishvili et al.,(16) Rhoads et al.,(142) Wright et al.(69)) demonstrate, however, that regardless of the phage delivery strategy used, phages are safe for use in phage therapy applications.
Summary
Recent trials of oral and topical phage therapy have shown the safety of phage therapy in humans. Experimental phage therapy has been reported to be efficacious in older studies and in a recent phase I/II trial; however, there have not been controlled clinical trials for aerosol phage therapy.
Conclusion
The current experience with phage treatment of bacterial infections indicates that it is likely that safe and efficacious phage therapeutics can be developed. Phages have the potential to become a much-needed tool in the fight against extensively antibiotic-resistant bacteria, including those causing respiratory infections. However, many pharmacokinetic questions remain open, and the ideal route of delivery for lung infections has not yet been determined. Although there is evidence to show the safety and efficacy of phage therapy, there is a real need for in vitro and in vivo studies specifically investigating aerosol phage delivery to the lung. Local administration of inhalable phages is an attractive prospect, as it may provide advantages, in particular for infections that are walled off from the systemic circulatory system, such as in CF patients.
Initial studies probing the feasibility of inhalable phage dosage forms appear promising, and compatibility with nebulization and dry powder delivery has been shown. Most chemistry, manufacturing, and controls questions, except for long-term storage stability, either have been addressed successfully or can be answered using the extensive development history with viral vaccine manufacturing.
The future of phage therapy in the West will depend on economic considerations as phage products move into more expensive late-stage clinical trials. Whether it will become attractive to industry to advance these much needed therapeutics to the commercial stage will be decided by patentability, market projections, potential financial incentives by governmental or nonprofit organizations, and by the prescribed regulatory pathway for these products. Currently, there are no specific regulatory guidelines for the development and approval of bacteriophage therapeutics in Europe and North America. Specific guidelines or clarifications regarding current applicable regulations would be desirable to remove development risks for sponsors. The regulatory framework should take into account that bacteriophage strains will likely have to be updated rapidly and may have to be adjusted to locally prevalent bacterial strains,(144) similar to the modifications required in the case of attenuated viral influenza vaccines. For this reason, a standard formulation and delivery system for bacteriophages that can be proven to be generally safe and suitable for a wide variety of interchangeable phage strains appears highly desirable.
Footnotes
Author Disclosure Statement
The authors declare that no conflicts of interest exist.