
Abstract
Abstract
Parenteral IFN-γ was unsuccessful as a treatment for pulmonary fibrosis. Inhaled IFN-γ targeted to the lungs may be more effective. Our patient, a 56-year-old male with biopsy proven usual interstitial pneumonia (UIP) and declining pulmonary function tests (PFTs) was initially diagnosed with idiopathic pulmonary fibrosis (IPF). He enrolled in a 2-year research protocol and was treated with inhaled IFN-γ (100 μg, Actimmune, Horizon Pharma, Deerfield, IL) 3 times per week. After completion of the protocol, he was able to secure the drug and continued therapy for a total of 7 years. He felt better, returning to work. His only complaint was transient cough during inhalation. PFTs improved (e.g., DLCO, 58% at baseline, 81% at 2 years, 69% currently). Clinical monitoring showed preserved exercise tolerance and stable CT scans. He was ultimately diagnosed (year 5) with scleroderma-like connective tissue disease after he developed sclerodactyly and a positive antinuclear antibody. Inhaled IFN-γ was well tolerated for 7 years and may stabilize fibrotic lung disease.
Introduction
F
Case Report
A 56-year-old male never smoker presented with one year of progressive shortness of breath. He denied cough or sputum production. He had difficulty breathing at work, complaining of 1–2 flight dyspnea. Review of systems was positive for Raynaud's phenomenon and shoulder pain. There were no reports of fever, chills, night sweats, headaches, wheezing, hemoptysis, dry mouth, dry eyes, dysphagia, or rash.
Past medical history included gastroesophageal reflux disease (GERD), osteoarthritis, atrial fibrillation, and hypothyroidism. Surgical procedures included appendectomy and tonsillectomy as a child, and arthroscopic shoulder surgery as an adult. Family history was significant for a sister with myasthenia gravis. Medications included levothyroxine, metoprolol, sildenafil, and omeprazole. At presentation, he was employed as a plumber. In his 20s he worked as a welder and machinist in an automotive plant. He reported no exposures to birds.
On physical exam, he was well-developed, well-nourished, and without respiratory distress. Skin and mucous membranes were unrevealing. Auscultation of the chest revealed fine bibasilar crackles. There were no appreciable abnormalities on cardiac exam.
As part of his initial evaluation, blood work was sent for connective tissue disorders. All tests were negative, including anti-nuclear antibody (ANA), rheumatoid factor (RF <12), C reactive protein (CRP = 10), erythrocyte sedimentation rate (ESR = 7), and anti-SCL70 antibodies (<0.2).
Computed tomography (CT) of the chest demonstrated ground glass and reticular opacities at the bases (Fig. 1). Honeycombing was not seen and he was referred for open lung biopsy (Fig. 2). Pathology reviewed at New York University was officially reported as showing “patchy involvement of parenchyma by honeycomb-type dense fibrosis and fibroblastic foci predominantly in subpleural and peribronchiolar distribution, associated with bronchiolar metaplasia, fatty metaplasia, smooth muscle proliferation, and lymphoid aggregates.” These findings were consistent with usual interstitial pneumonia (UIP). In the absence of a discernible cause for this pulmonary fibrosis, despite an exhaustive evaluation at an established pulmonary fibrosis center, he was diagnosed with idiopathic pulmonary fibrosis (IPF).
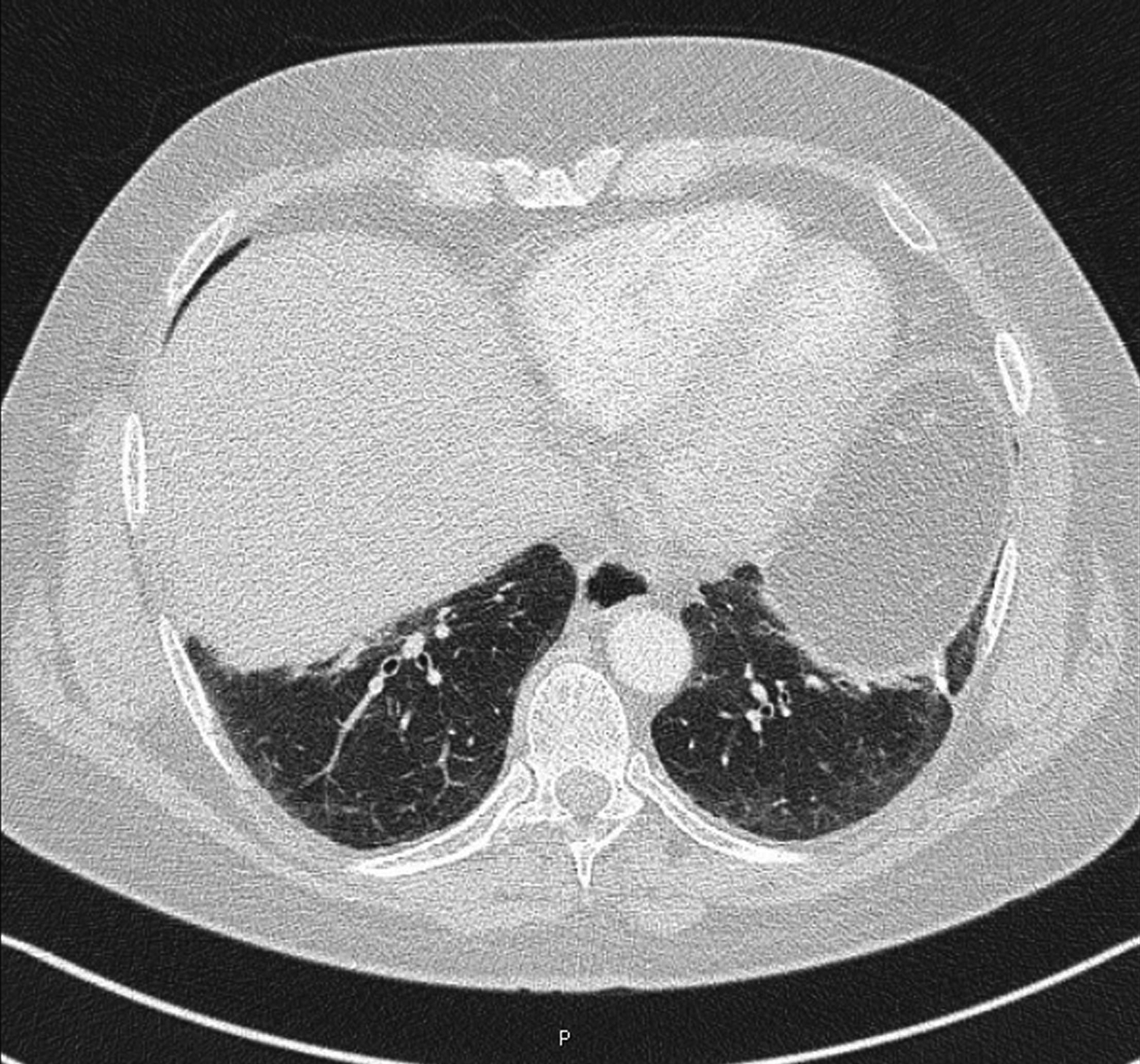
Noncontrast CT of the chest performed in 2006 demonstrating ground glass opacities at the time of initial presentation. Subsequent studies showed no significant change.
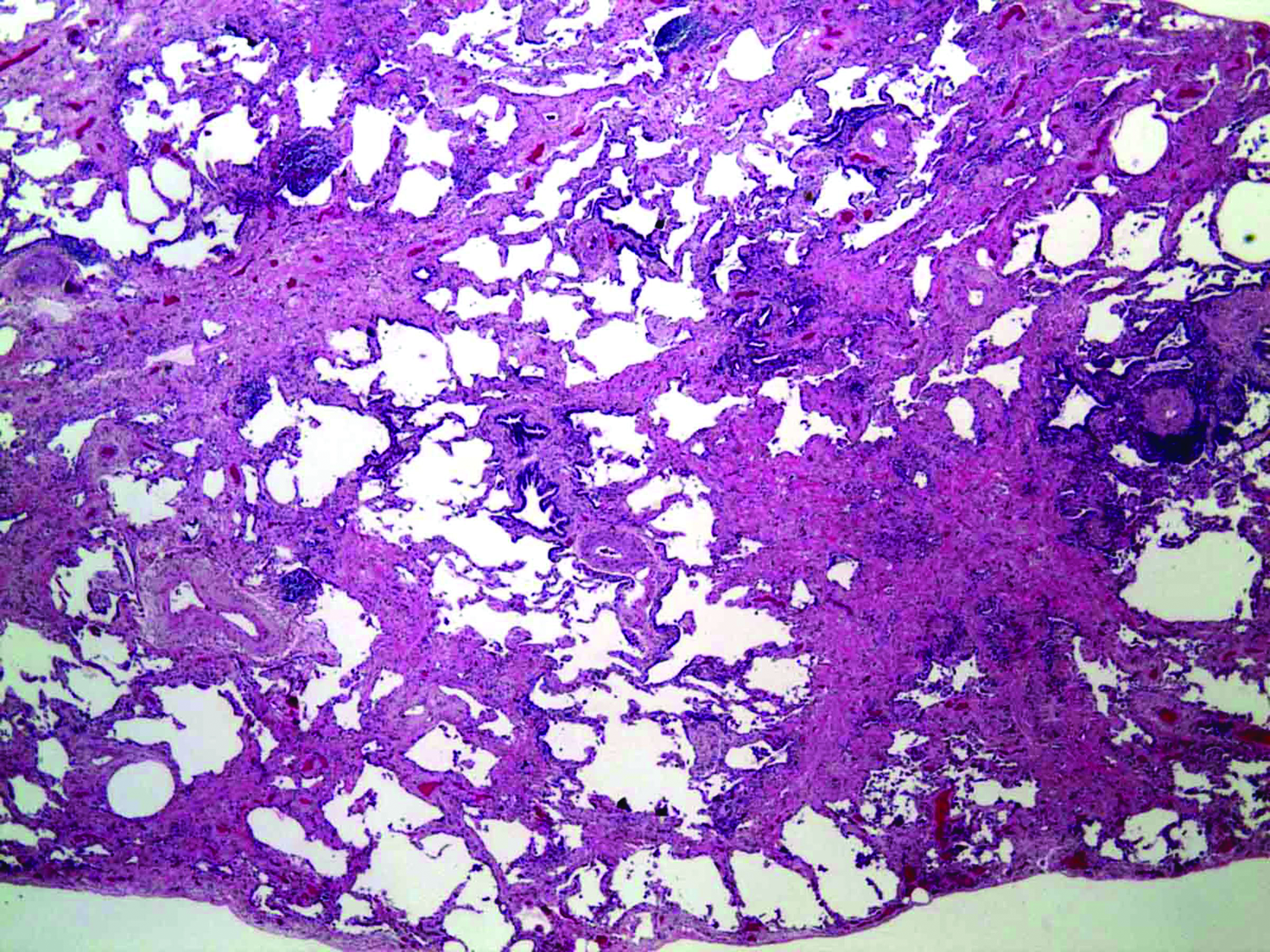
Open lung biopsy performed in 2006 with hematoxylin and eosin stain at 20x magnification showing temporal heterogeneity consistent with UIP.
The patient was enrolled in an experimental trial of inhaled IFN-γ.(4) Prior to initiation of inhaled IFN-γ, all patients had been observed clinically for 4–6 months. During this time, routine serial pulmonary function tests (PFTs) were obtained. For example, our patient was followed for 5 months. His PFTs demonstrated a steady decline in total lung capacity (TLC) and diffusion capacity of the lung for carbon monoxide (DLCO). For example, DLCO fell from 75% to 58% predicted (Fig. 3), a finding consistent with active pulmonary fibrosis.

PFT data over time, 0 % indicates PFT at start of inhaled IFN-γ, points are actual data, lines generated by smoothing. The y-axis represents the percentage change from baseline, defined as start of inhaled IFN-γ. Observations before baseline demonstrate decreasing TLC (■) and DLCO (•). Forced Vital Capacity (FVC;○) stabilized after start of therapy.
Then he received 100 μg of Actimmune (Horizon Pharma, Deerfield, IL), three times per week via vibrating mesh nebulizer [I-neb, Philips Respironics, Parsippany, NJ, from 2007–2010, Omron U22, Omron Healthcare Inc., Bannockburn, IL from 2011–2014]. No other treatments were prescribed except sildenafil for Raynaud's. During therapy with inhaled IFN-γ, PFTs improved with the greatest change in DLCO, which increased to 81% at 1.5 years of therapy. The dose of deposited drug in his lungs was measured on two occasions by gamma camera (67 and 91.8 μg).(4) The experimental protocol concluded after 1.5 years of therapy but he was able to secure the drug and continued treatment without interruption for a total of 7 years. His most recent DLCO was 69%.
After 5 years of treatment, the patient complained of worsening Raynaud's and GERD and was found to have a positive ANA. However, anti-SCL70 antibodies and CRP were persistently negative. Referral was made to rheumatology, where a diagnosis of scleroderma-like connective tissue disorder was made and treatment with hydroxychloroquine started. His symptoms improved. Repeat serologies were all negative with no serologic evidence of scleroderma or other connective tissue disease. Therapy with inhaled IFN-γ continued throughout his evaluation and treatment with hydroxychloroquine.
A right heart catheterization in 2009 did not show evidence of pulmonary hypertension (pulmonary artery pressure <25 mmHg). Learning of the prognosis of IPF after his initial diagnosis, he quit his job and put his affairs in order. However, he has done so well that he has resumed work, now as a travel agent. Throughout the course of his 7 year follow up, serial CT scans of the chest showed stability of previously mentioned radiographic findings.
Discussion
Our patient's experience with inhaled IFN-γ is encouraging. The typical decline in pulmonary function associated with fibrotic diseases of the lung, idiopathic or otherwise related to systemic disorders, seemed to reverse direction and stabilize. Additionally, the medication itself was well tolerated over a prolonged course with the patient only reporting cough at the onset of inhalation that resolved as the treatment progressed. In general, over 7 years he remained well, reporting only minor respiratory infections.
An important concern with inhaled medications, particularly biologics, is pulmonary toxicity. In our studies of patients with TB and IPF, treated with inhaled IFN-γ, there was no evidence of pulmonary or systemic toxicity.(3,4) In addition, for inhaled or systemic IFN-γ there has been no reported association with scleroderma-like symptoms.(1–4) Inhaled IFN-γ avoids the well-established side effects of systemic interferon.(1–4) The latter include fever, headache, myalgia, arthralgia, fatigue, ecchymosis, petechiae, and gastrointestinal bleeding.
In addition to the more favorable side effect profile, inhaled IFN-γ results in a far higher concentration of IFN-γ in the bronchoalveolar lavage (BAL) fluid than that following systemic interferon.(4,5) A 2004 study of 17 patients receiving 200 micrograms of subcutaneous IFN-gamma three times per week over the course of 6 months did not show a statistically significant increase of IFN-γ in BAL fluid.(5) However, the concentration of IFN-γ in BAL fluid, when given as 100 μg inhaled 3 times weekly, was several orders of magnitude higher.(4)
In late 2014, pirfenidone and nintedanib were approved for treatment of IPF.(6,7) While pirfenidone's mechanism of action is not clearly understood, it is speculated that it reduces fibroblast proliferation and production/activity of TGF-beta.(8) Nintedanib, while primarily a tyrosine kinase inhibitor and working mostly via PDGF and VEGF, has additionally been shown to block TGF-beta induced transformation of fibroblasts to myofibroblasts, albeit through SMAD independent pathways.(9) IFN-γ, a potent inhibitor of TGF-β offered promise as a treatment modality, but was ineffective parenterally in large clinical trials.(1,2) However, when targeted directly to the lungs, it is a potent immunomodulator in normal subjects(10) and patients with tuberculosis.(3,11)
Limited conclusions can be drawn from our experience with a solitary patient. Determining cause and effect is impossible. Whether the inhaled drug stabilized lung function is also speculative. However, this patient's experience is encouraging and combined with other studies, we believe the utility of inhaled IFN-γ for the long-term treatment of fibrotic pulmonary disorders warrants further study.
Footnotes
Acknowledgments
The authors would like to thank Lorraine Morra for assistance in manuscript preparation.
Funding: No funding or grants supported this case report.
Author Disclosure Statement
Stony Brook University and New York University jointly hold patents in the treatment of IPF with inhaled IFN-γ (Dr. Smaldone and Dr. Condos). Dr. Fusiak reports no potential conflicts of interest.