
Abstract
Abstract
Background:
Fluticasone propionate/formoterol (FP/FORM) is a pressurized metered-dose inhaler (pMDI; Flutiform®) approved for use in adolescents and adults and under development for pediatric use.
Objective:
To compare short-term growth in asthmatic children treated with FP/FORM, FP pMDI with valved holding chamber, and beclomethasone dipropionate (BDP) in a breath-actuated device.
Methods:
Children with persistent asthma (n = 48; 5 to <12 years) participated in an assessor-blinded, randomized, three-way crossover trial with run in, wash out, and active treatment periods, each of 2 weeks duration. Interventions were FP/FORM 100/10 μg b.i.d. with an AeroChamber Plus® Flow-Vu® Spacer, FP pMDI (Flixotide®) 100 μg b.i.d. with a Volumatic® spacer, and extra-fine BDP breath-actuated inhaler (Aerobec®/QVAR® Autohaler®) 100 μg b.i.d. Lower leg growth rate (LLGR) was measured by knemometry.
Results:
The least square (LS) mean difference in LLGR between FP/FORM and FP (per protocol population) was −0.006 mm/week (95% CI: −0.095 to 0.084; p < 0.001 for noninferiority [noninferiority margin −0.200 mm/week]). Both treatments elicited no change from baseline off-treatment growth rate. The LS mean treatment difference of FP/FORM versus BDP was 0.116 mm/week (95% CI: −0.004 to 0.235; p = 0.057) and of FP versus BDP 0.163 mm/week (95% CI: 0.078–0.249; p < 0.001). Results in the full analysis population were: FP/FORM versus FP −0.012 mm/week (95% CI: −0.080–0.056; p < 0.001); FP/FORM versus BDP 0.143 mm/week (95% CI: 0.064–0.222; p < 0.001); FP versus BDP 0.163 mm/week (95% CI: 0.093–0.233; p < 0.001).
Conclusions:
FP/FORM pMDI with AeroChamber and FP pMDI with Volumatic spacer did not affect lower leg growth, measured by knemometry, in asthmatic children. Conversely, extra-fine BDP from a breath-actuated inhaler resulted in short-term growth suppression.
Introduction
I
Furthermore, the devices delivering the ICS and the pharmaceutical characteristics of the drug formulation (e.g., conventional versus extra-fine particle size formulation, aerosol plume character, etc.) are relevant as they affect the relative extent of, and balance between, pulmonary and gastrointestinal drug deposition. Alterations in pulmonary drug deposition and/or the fraction of the drug absorbed through the gastrointestinal tract (GIT) may in turn alter the systemic drug load and associated systemic effects.(6,7) Thus, even when well-known ICS are developed in novel formulations (e.g., per the switch from chlorofluorocarbon to hydrofluoroalkane propellants), or in novel devices, it is prudent to robustly evaluate their potential for systemic effects.(7)
The growth-suppressive potential of inhaled corticosteroids can be assessed over a few weeks by using knemometry, which has been validated as a sensitive, highly reproducible and noninvasive technique to measure short-term lower-leg growth.(6) Although knemometry studies cannot accurately predict the extent of any treatment effect upon final adult height, the absence of an effect on short-term lower-leg growth suggests that an adverse effect upon growth is unlikely with long-term use.(6)
A fixed-dose pressurized metered-dose inhaler (pMDI) combination of fluticasone propionate and formoterol fumarate dihydrate (FP/FORM; Flutiform®; Napp Pharmaceuticals Limited, Cambridge, United Kingdom) has been approved for the maintenance treatment of asthma in adults and adolescents(8) in over 30 countries within Europe, Asia Pacific, and elsewhere. The present study was conducted to assess short-term lower leg growth in asthmatic children treated with FP/FORM as compared with FP alone and to extra-fine particle beclomethasone dipropionate (BDP). We hypothesized that a short-term growth-suppressive effect would be observed with beclomethasone, but not fluticasone-based treatments at equivalent doses, in view of the substantial oral bioavailability of the former.
Materials and Methods
The study was conducted in accordance with the International Guidelines issued by the European Commission in 1990 and the Declaration of Helsinki and approved by the Local Ethics Committee.(9) The International Conference on Harmonization for Good Clinical Practice Guideline was followed.(9) Case numbers were: Danish Health and Medicines Authority: 2013112520, and Ethics Committee Region Midtjylland: 1-10-72-355-13.
Patients
Inclusion criteria were: male and female prepubertal (Tanner stage 1 of puberty development)(10) children aged 5 to 11 inclusive, with a diagnosis of asthma; normal growth development (height between 3rd and 97th percentiles according to Danish standard height growth charts); current use of either short-acting β2—agonist and/or non-ICS controller medications (e.g., cromones or leukotriene receptor antagonists) at a stable dose for at least 3 months; no ICS for at least 2 weeks; ability to correctly use a peak-flow meter and to perform lung function testing; satisfactory pMDI plus spacer and Autohaler inhaler technique; willing to comply with all study requirements; and written informed consent from both parents/guardians with assent from the child before any trial procedure was carried out. Exclusion criteria were: concomitant severe diseases and diseases which the investigator believed could affect study outcome measures, including malignancies; chronic systemic or lung diseases, for example, cystic fibrosis; any asthma exacerbation of any severity during 3 months preceding visit 1; hospitalization for asthma within 6 months before visit 1; current or recent (within 3 months before visit 1) systemic (oral, parenteral, or depot) corticosteroid therapy; continuous use of ICS for asthma symptoms; a clinically significant respiratory tract infection within 4 weeks of visit 1; any fracture in the leg to be measured by knemometry within 6 months before visit 1; no major surgery requiring general anesthesia within 3 months before visit 1; no febrile illnesses with a temperature greater than 39°C within a week of visit 1; known or suspected hypersensitivity to any of the treatments used in the study; the use of concomitant medications liable to alter the pharmacokinetics or pharmacodynamics of study treatments, for example, CYP 3A4 inhibitors, such as ketoconazole or β-blockers; and participation in an investigational drug trial within 30 days preceding visit 1.
Furthermore, forced expiratory volume in 1 second (FEV1) was to be ≥80% of the value at screening. A subsequent decrease in pulmonary function by 20% or more of the screening value would lead to exclusion from the study, to avoid the potential confounding influences of reduced pulmonary drug deposition, or worsening asthma control upon growth rate.
Study design
The study was a randomized, assessor-blinded, crossover trial with a run in, two wash out, and three treatment periods, each of 2 weeks duration(6) (Fig. 1). At the first (screening) visit, the child and his/her parents were informed about the study and consent, and assent to participate were obtained. Eligibility was evaluated, and demography and pubertal development using the Tanner staging system were recorded by a doctor (O.D.W.) if the patient was eligible.(10) At the second visit, patients were randomized. The total study duration for each patient was 12 weeks.
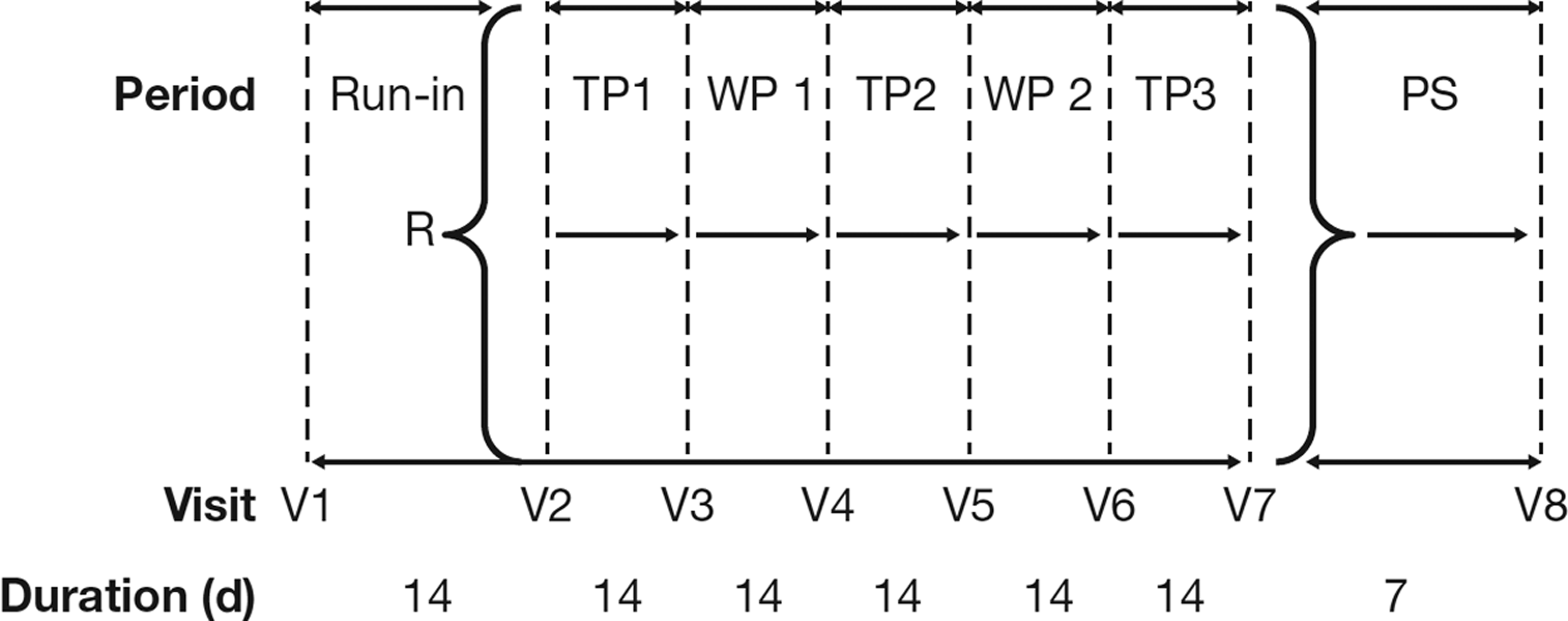
Study design. d, days; PS, poststudy; R, randomization; TP, treatment period; V, visit; WP, washout period.
Patients attended the clinic on the first and last day of each period at the same time in the afternoon between 13:00 and 19:00 ± 1 hours.(6) Knemometry and pulmonary function tests were performed at all visits. The knemometer was set up according to standard methodology and the right lower leg measurement was taken by a single trained assessor who was blinded to study treatment. The study was conducted at The Children's Clinic Randers, Randers, Denmark, from February to May 2014.
Active treatments were as follows, each administered as two puffs in the morning and two puffs in the evening:
• Treatment A: FP/FORM (suspension) pMDI 50/5 μg (Flutiform) through the AeroChamber Plus® Flow-Vu® Anti-Static Spacer (149 mL anti-static polymer; Trudell Medical International, Ontario, Canada). Mass median aerodynamic diameter (MMAD) 3.2 μm.(11) • Treatment B: FP (suspension) pMDI 50 μg (Flixotide® Evohaler® [Glaxo Wellcome S.A., Burgos, Spain]) through the Volumatic® Spacer [750 mL nonconducting polymer; GlaxoSmithKline, Uxbridge, United Kingdom]. MMAD 2.6 μm.(12) • Treatment C: BDP (solution) breath-actuated inhaler 50 μg (Aerobec® Autohaler® [3 M Healthcare Ltd., Loughborough, United Kingdom]). MMAD 1.2 μm.(13)
Spacers for Treatments A and B were washed in lukewarm detergent solution and left to drip dry for 24 hours before use.
The children were randomized to one of six treatment sequences (ABC, ACB, BAC, BCA, CAB, or CBA), determined by means of a computerized randomization scheme prepared in balanced blocks, provided by Mundipharma Research Limited (Cambridge, United Kingdom). The children took the medications at 07:00 hours and 19:00 hours ±60 minutes. Mouth rinsing was performed after inhalation. No asthma drugs other than the study medication, as-needed albuterol breath-actuated inhaler, oral antihistamines, and oral leukotriene receptor antagonists, were allowed during the study period. Compliance with study drug administration was assessed by weighing the pMDIs immediately before they were dispensed to participants and at the end of the study. Compliance rates (actual used weight/expected weight loss × 100%) outside of the range of 75%–125% were deemed protocol deviations, with rates outside 65%–135% deemed major and leading to exclusion from the per protocol population.
All lower leg length measurements (the distance from the surface of the right flexed knee to the bottom of the sole) were performed by the same trained assessor without reference to previous recordings. At each visit, four independent estimations were made and the first was discarded.(6) The children were sitting 10 minutes before and refrained from physical activity on the days the measurements were made. LLGR was calculated for each period by the conventional method as previously described(14): the change in lower leg length in a given period divided by the duration of the period expressed in mm/week.
For clinical monitoring purposes, peak expiratory flow (PEF) was measured at home in the morning and evening (best of three) using the electronic Peak Flow Meter (Vitalograph® Asma-1 device; Vitalograph (Ireland) Ltd., Co Clare, Ireland) before intake of study medications. FEV1 was measured at each visit using a Vitalograph® spirometer (Spiropharma, Copenhagen, Denmark). Asthma exacerbations requiring intensified treatment with systemic or inhaled glucocorticoids or other medications would lead to withdrawal from the study.
Statistics
The study was powered to demonstrate noninferiority between lower leg growth rates (LLGRs) during treatment with FP/FORM 100/10 μg and FP 100 μg twice daily (b.i.d.).(15) For the sample size calculation, a standard deviation (SD) of the period differences of 0.30 mm/week and a treatment difference of 0 mm/week between the FP/FORM and the FP treatment was assumed. The SD was based on observations in previous knemometry studies of inhaled corticosteroids.(6) A noninferiority margin of −0.200 mm/week was used, based on an estimated placebo growth rate of 0.40 mm/week, the observation that >25% reduction in short-term LLGR translates to a reduction in medium-term growth rate of between 0.5 and 1.5 cm/year, and the technical error margin of 0.10 mm associated with knemometry.
Thirteen pairs of evaluable subjects per treatment comparison were required to achieve 90% power to show noninferiority. Assuming that ∼40% of the randomized subjects were likely to be excluded from the per protocol population, a minimum of 48 subjects (eight per treatment sequence) were needed for randomization to achieve 26 subjects in the per protocol analyses.
Analysis populations were defined as the enrolled population: all children for whom informed consent and assent were obtained; the randomized population: all children who were randomized to a treatment sequence; the full analysis population (FAP): all randomized children who received at least one dose of investigational medicinal product and had a valid baseline and at least one valid postbaseline LLGR (i.e., this is a modified intent-to-treat population); and the per-protocol population (PPP): all FAP children without major protocol deviations. Baseline LLGR for each treatment was defined as the LLGR in the off-treatment period (i.e., run-in or washout period) immediately preceding the treatment period in question. A valid LLGR was defined as being within the range of −0.1 to 1 mm/week.(6)
The least square mean (LS Mean) difference (and associated 95% CI) between short-term LLGRs on FP/FORM and FP was estimated using an analysis of covariance (ANCOVA) with fixed terms for treatment, period, and treatment sequence; subject within sequence as a random effect, baseline growth rate, and % predicted FEV1 as covariates. Only data from subjects providing data for both treatments were included in the analysis. Noninferiority of FP/FORM compared with FP (the primary study comparison) was concluded if the lower limit of the 95% CI for the LS mean difference was greater than or equal to −0.2 mm/week. This was equivalent to a one-sided test at a 2.5% level of significance. The primary analysis population was the PPP, whereas a sensitivity analysis was performed on the FAP. In addition, mean LLGR during the FP/FORM and FP periods, respectively, was compared with mean LLGR during the BDP period.
The same ANCOVA model used for the primary comparison (FP/FORM vs. FP) was employed to estimate LS mean differences and associated 95% CIs for these secondary comparisons and corresponding p values from a two-sided test, testing the hypothesis of no difference between treatments. Finally, LLGRs during the FP/FORM, FP, and BDP periods were compared with the LLGR during the off-treatment period (run in or wash out) immediately before the respective treatment period as a post hoc analysis. These comparisons were made using simple two-sided t-tests, testing the hypothesis of no change in LLGR under treatment. All analyses were performed after study completion and database lock. Statistical analyses were performed using SAS® version 9.3 or higher (SAS Institute, Cary, NC).
Results
All children were recruited from a secondary referral center. There were no screening failures (Fig. 2). The randomized study population comprised 38 boys and 10 girls, 45 Caucasians, 1 Black, and 2 Asians, with a mean age of 8.7 years (range 5–11 years; SD 1.65). Mean height was 135.5 cm [range 111–157 cm (SD 9.59)], mean weight 32.7 kg [range 20–74 kg (SD 9.35)]. All children were prepubertal (Tanner stage 1 of pubertal development). Mean asthma duration was 5.8 years (range 3 months–11.3 years). Eight children were allocated to each of the six treatment sequences. For personal reasons two children withdrew consent after receiving both the FP/FORM and FP treatments, but without receiving BDP.
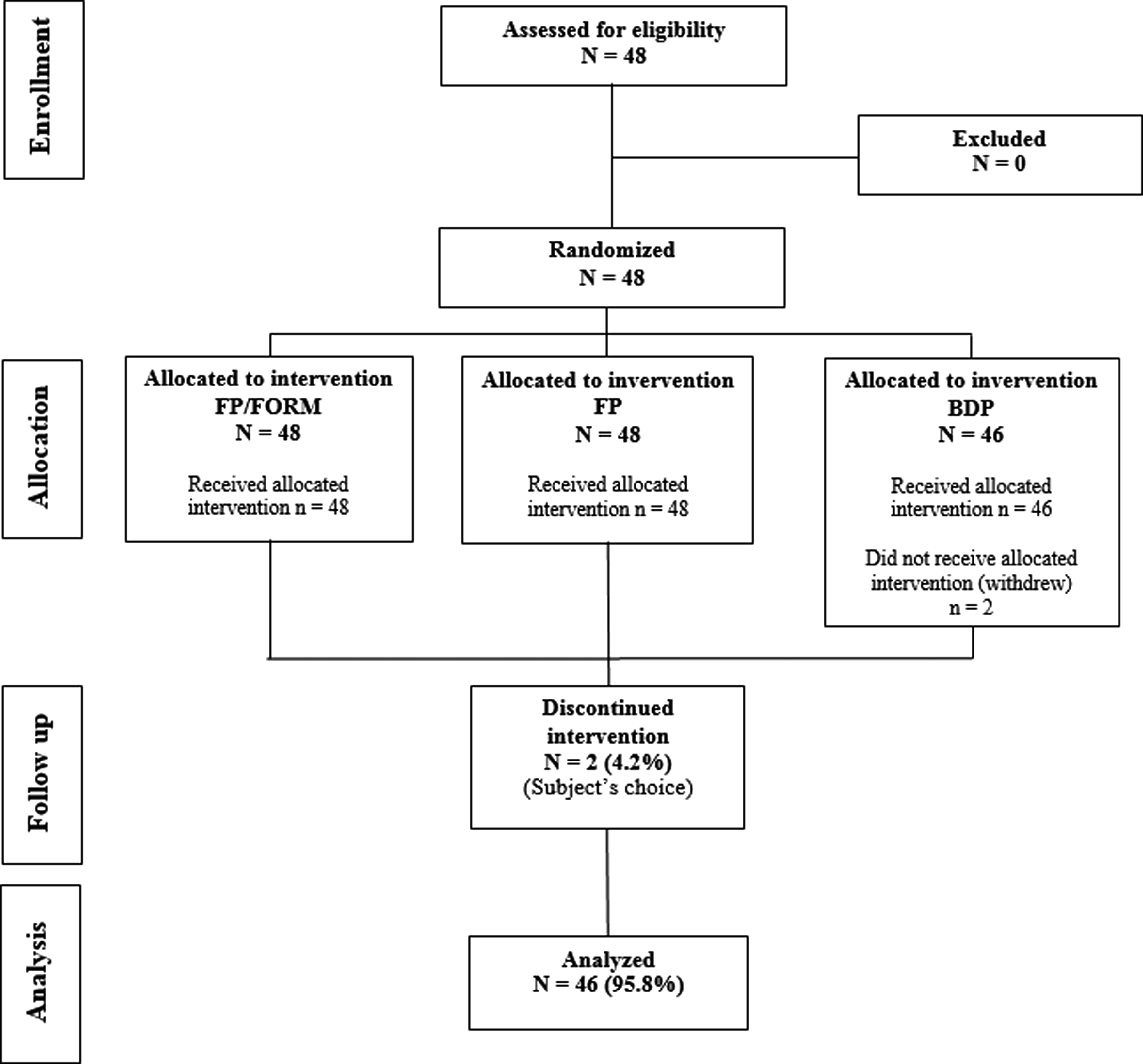
Subject entry and participation in the study. BDP, beclomethasone dipropionate; FP/FORM, fluticasone propionate/formoterol fumarate.
Another child did not have a valid baseline LLGR value for either the FP/FORM or the FP periods, and was, therefore, not included in the FAP for those two treatments. Thus, the FAP consisted of 47 children for the FP/FORM and FP periods, and 46 children for the BDP period. The PPP consisted of 31 children for the FP/FORM period, 37 for the FP period, and 35 for the BDP period. Reasons for exclusion from the PPP are given in Table 1.
Evaluable data for at least two treatment groups were required for inclusion in Total Per Protocol population. Subjects could be excluded from the PPP for more than one reason.
BDP, beclomethasone dipropionate; FP/FORM, fluticasone propionate/formoterol; IMP, investigational medicinal product; N, number of subjects in randomized population; n, number of subjects in specified category; %, percentage based on N.
Parallel boxplots of LLGR by treatment are presented in Figure 3, and inferential statistics for the change in LLGR between the preceding off-treatment baseline and subsequent treatment periods are presented in Table 2 for the PPP. The results of the primary outcome measure showed that the LS mean treatment difference for FP/FORM versus FP was −0.006 mm/week (95% CI: −0.095, 0.084 mm/week, p < 0.001). Noninferiority was therefore proven as the lower limit of confidence interval, and the difference was greater than the prespecified noninferiority margin of −0.200 mm/week. A similar result was observed for the FAP (LS mean difference −0.012 mm/week [95% CI: −0.080 to 0.056, p < 0.001]). The technical error of the knemometer (i.e., the SD of each measurement) was 0.14 mm.
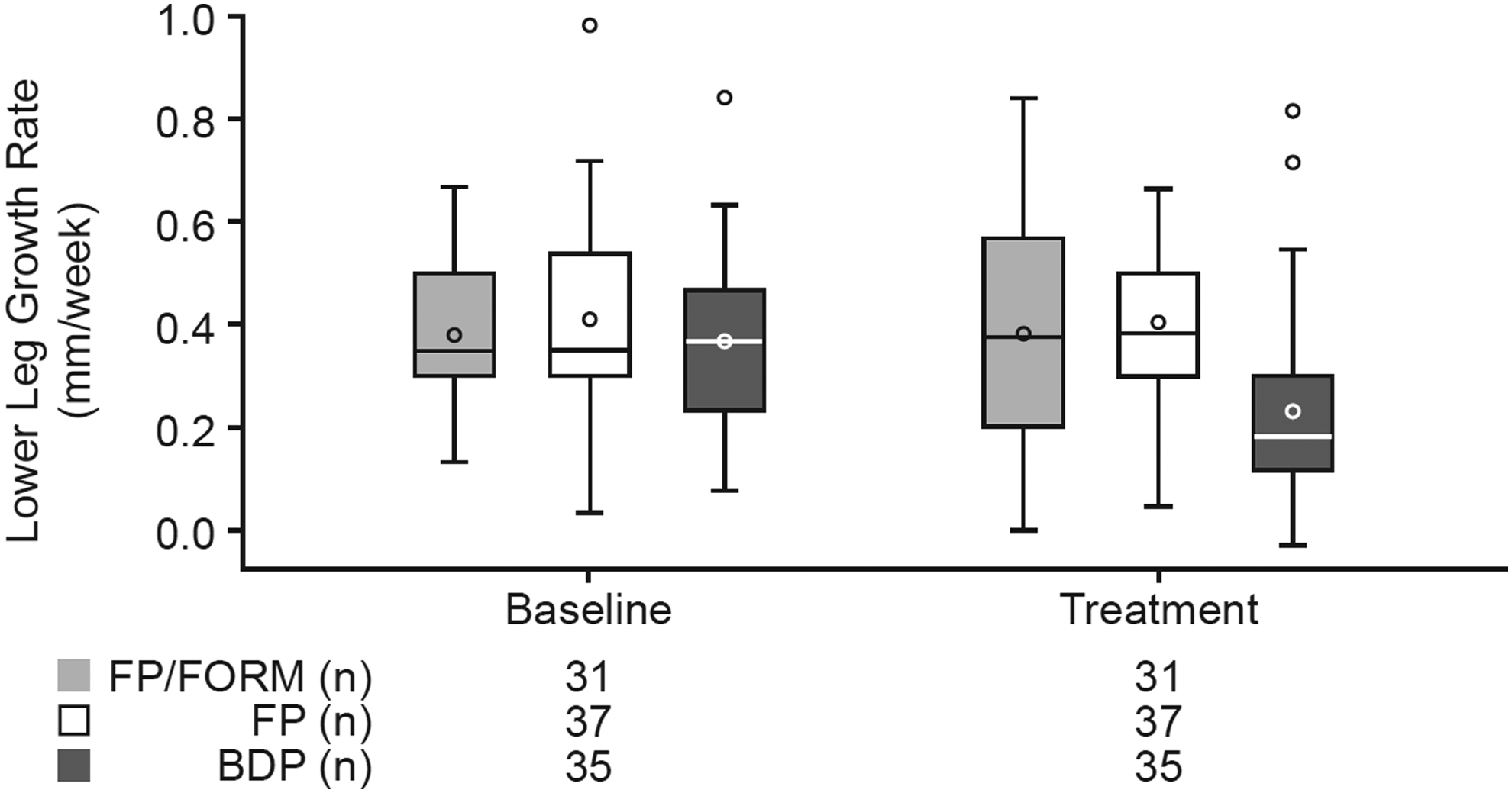
Parallel boxplots for lower leg growth rate for the per protocol population during baseline off-treatment and treatment periods. Baseline is defined as the value from the nontreatment period (run-in or washout) preceding the respective treatment period. The box represents the IQR (25th and 75th percentiles). The line inside the box represents the median and the o inside the box represents the mean. The whiskers represent the maximum value below 1.5 × IQR above 75th percentile and the minimum value above 1.5 × IQR below 25th percentile. The symbols outside the whiskers represent outliers. IQR, interquartile range.
Baseline is defined as the value from the nontreatment period (run-in or washout) immediately preceding the respective treatment period.
Secondary comparisons of LLGR showed the LS mean treatment difference of FP/FORM versus BDP was 0.116 mm/week (95% CI: −0.004 to 0.235 mm/week, p = 0.057) and of FP versus BDP 0.163 mm/week (95% CI: 0.078 to 0.249 mm/week, p < 0.001). Similar results were seen in the FAP: LS mean treatment difference of FP/FORM versus BDP was 0.143 mm/week (95% CI: 0.064 to 0.222 mm/week, p < 0.001) and FP versus BDP 0.163 mm/week (95% CI: 0.093 to 0.233 mm/week, p < 0.001). Finally, when LLGRs on active treatments were compared with baseline LLGR, based on the PPP, group means were not reduced during FP/FORM (change from baseline 0.003 mm/week; p = 0.950) or FP (−0.004 mm/week; p = 0.915) periods, whereas BDP was associated with a statistically significant suppression (−0.137 mm/week; p = 0.006), representing a 37.1% reduction in group mean LLGR.
Only minor and clinically irrelevant variations in group means of FEV1 (Table 3), PEFR, and use of inhaled Airomir Autohaler, which would not be expected to confound comparisons of LLGR, were recorded.
Baseline is defined as the value from the nontreatment period (run-in or washout) preceding the respective treatment period.
Mean morning FEV1 is calculated as the mean from all morning dose FEV1 measurements in that period recorded through the peak expiratory flow (PEF) meter.
FEV1, forced expiratory volume in the first second.
Nine (18.8%) children experienced 12 treatment-related adverse events during the study: 4 (8.3%) during FP/FORM, 3 (6.3%) during FP, and 3 (6.5%) during BDP treatment. Cough was the most common adverse event experienced by 4 (8.3%) children: 1 (2.1%), 2 (4.2%), and 1 (2.2%) during the FP/FORM, FP, and BDP periods, respectively. All events were mild and resolved before termination of the study. There were no serious adverse events.
Discussion
This study was performed to support the development of FP/FORM for use in asthmatic children. It was hypothesized that FP/FORM and FP would not alter LLGR at the doses employed,(16) despite administration of both pMDI treatments through spacers (per GINA recommendations(1)) known to increase the pulmonary drug delivery of these respective formulations (hence their systemic effect potential) compared with administration through a pMDI alone.(17,18) FP pMDI was chosen as the primary comparator as it contains the same corticosteroid component as the FP/FORM pMDI, represents the same pharmaceutical device, and the benefit:risk of FP in pediatric asthma is long established.(19) To lend rigor to this primary noninferiority comparison, we included a third treatment arm(20) in the study as a positive control, BDP from the Aerobec Autohaler (also known as QVAR® Autohaler).
While the pulmonary anti-inflammatory effects of these three treatments would be expected to be similar, we anticipated that marked differences in oral bioavailability between FP (which exhibits <1% oral bioavailability(21)) and BDP (40% oral bioavailability(21)) could increase the propensity to see differences in LLGR suppressive effects between these ICS; since systemic exposure to inhaled drugs depends on drug absorption through both pulmonary and gastrointestinal routes.
To simplify treatment and limit the potential for confusion and device handling errors for participants, the present study was single (assessor-) blind. While this is a potential limitation, it was not anticipated to lead to bias due to the highly objective nature of the key endpoints. Previous studies with similar designs have confirmed the robustness of a single-blind knemometry design.(7) The study team, including persons involved in conducting the analysis, remained blinded to the treatments, which the children were randomized to until after database lock.
A further potential limitation of our study is that different devices were used to deliver different treatments. However, our study was intended to compare treatments in their approved form rather than to evaluate active substances per se. Thus, FP/FORM was used in conjunction with the AeroChamber Plus Flow-Vu Anti-Static Spacer, since this spacer was used in a pediatric efficacy study also intended to support the regulatory approval of FP/FORM.(22) FP was used with the Volumatic spacer with which it is recommended for use.(19) The finding of noninferiority between FP/FORM and FP is unlikely to have been confounded by the use of different spacers since a comparative knemometry trial of beclomethasone pMDI delivered through the AeroChamber Plus and the Volumatic showed noninferiority of LLGRs,(23) as a previous adrenal axis study of fluticasone/salmeterol pMDI showed similar cortisol-suppressive effects through the AeroChamber Plus and Volumatic spacers,(24) and since in vitro data support similar performance of the AeroChamber Plus and AeroChamber Plus Flow-Vu Anti-Static spacers for multiple different pMDI formulations.(25)
Note that in all these studies nonconducting spacers were either washed in detergent and dried,(23,25) as in the present study, or effectively primed through the use of multiple aerosol actuations.(24) With regard to the BDP Autohaler, this breath-actuated pMDI is widely approved for use in children of 5 years and above.
Data presented showed no short-term growth suppression with FP 100 μg b.i.d. from either the combination or the monotherapy pMDI, both with a spacer. However, a similar short-term knemometry study of FP pMDI (225 μg in the morning and 125 μg in the evening with the AeroChamber Plus spacer) showed a statistically significant 82% suppression of LLGR,(26) supporting previous observations of a dose-related effect of ICS on LLGR,(27) and indicating that the FP doses employed in our study were at the cusp of the steep part of the dose–response curve.
In contrast to FP, extra-fine BDP through the Autohaler was associated with prominent attenuation of LLGR. Systemic exposure to BDP and its metabolites with Autohaler use results from a large fraction of the drug deposited in the oropharynx (OP) and GIT(28) becoming systemically available,(21) allied to systemic exposure derived through the pulmonary circulation. Previous knemometry trials of extra-fine BDP pMDI with an AeroChamber Plus and Volumatic spacer (which largely abate gastrointestinal drug deposition, but which do not alter the lung dose of BDP compared with that delivered through a pMDI alone(29)) have also consistently found 100 μg b.i.d. to significantly suppress LLGR.(23,30,31)
Hence, it was unsurprising to see a LLGR effect through the Aerobec Autohaler 100 μg b.i.d. given the additional OP/GIT-derived systemic dose imposed through this breath-actuated device; although the present trial is the first to confirm the systemic activity of extra-fine BDP through the Autohaler using knemometry. Since suppression of short-term LLGR of 25% or more has been shown to imply a clinically relevant risk of height growth suppression of 1–2 cm during the first year of continuous twice-daily treatment,(6) the observation of a 31.7% reduction in LLGR during the BDP period should be anticipated to pose a similar risk. Considering these findings, it is relevant that there are no head-to-head studies comparing the systemic effects of extra-fine BDP delivered with a spacer compared with an Autohaler, and no long-term studies (i.e., >1 year's duration) measuring height or final adult height with extra-fine particle BDP through either device. Such studies are warranted.
Conclusion
In conclusion, the present study demonstrated that FP/FORM 100/10 μg b.i.d. was noninferior to FP 100 μg b.i.d. in terms of effects upon LLGR in children aged 5 to 11 years with mild asthma. Growth rates during extra-fine particle BDP treatment were reduced when compared with both FP/FORM and FP periods and, in contrast to BDP, both FP/FORM and FP elicited no or little change from baseline off-treatment growth rates. Comparisons of FP/FORM, FP, and extra-fine BDP at higher dose levels have not been undertaken to ascertain whether the relative effects seen in this study would remain the same. However, FP/FORM is not intended for use at higher doses in children.
Footnotes
Acknowledgments
The authors would like to thank study participants, study nurse Signe Dreier for taking the knemometry measurements, and nurse Anne Karina Kjaer for help with recruiting children. This study was funded by Mundipharma Research Limited. EudraCT number 2013-004719-32, ClinicalTrials.gov identifier: NCT02063139.
Trade Mark Statements
®Flutiform is a registered trade mark of Jagotec AG. AeroChamber Plus and Flow-Vu are registered trademarks of Trudell Medical International. Flixotide Evohaler and Volumatic are registered trademarks of Glaxo Group Limited. Aerobec is a registered trade mark in Europe of either Meda AB or Teva Pharmaceuticals International GmbH. QVAR and Airomir are registered trademarks in Europe of either Ivax International GmbH, Teva Pharmaceuticals International GmbH, or 3M Company. Autohaler is a registered trade mark of 3M Company.
Author Disclosure Statement
O.D.W. was the Principal Investigator for this study. A.M. and S.D. were full-time employees of the study sponsor, Mundipharma Research Ltd., and Sabine Mersmann was a full-time employee of Mundipharma Research GmbH & Co. KG. The authors have no other financial relationships relevant to this article to disclose.