
Abstract
Abstract
Background:
Inhalation drug delivery is a fast, effective, and safe route of delivering medication directly to the lungs. Thanks to the large surface area and highly vascularized epithelium in lung, pulmonary drug delivery has been considered as an effective route to deliver drugs to the systemic circulation. Pirfenidone (PF), an oral antifibrotic agent, has been shown to slow down the progression of the lung fibrosis. Inhalation or intrapulmonary delivery of PF appears to be a good alternative to optimize drug delivery and minimize the dosage, adverse and nonspecific effects.
Methods:
Pulmonary fibrosis was induced by paraquat in rats. After induction of fibrosis, PF was administered via oral and inhalation routes for 14 consecutive days. The efficacy of oral and inhalation routes were compared by evaluating morphological changes, hydroxyproline content, tissue oxidative stress parameters, and proinflammatory and profibrotic genes expression including transforming growth factor beta 1 (TGF-β1), tumor necrosis factor alpha (TNF-α), tissue inhibitor of metalloproteinase 1 (TIMP-1), and matrix metalloproteinase 2 (MMP-2) genes.
Results:
The results showed similar therapeutic effects and efficacy for both inhalation and oral routes; however, the dose of inhalation route was much less than that for oral administration.
Conclusion:
In conclusion, PF offers great potential as an inhalation delivery formulation for treatment of pulmonary fibrosis.
Introduction
I
Pirfenidone (PF) is an orally active small molecule that exhibits potent antifibrotic and anti-inflammatory activities. It has been shown that it slows down the progression of lung fibrosis. In addition, the drug has been evaluated in large clinical trials for the treatment of idiopathic pulmonary fibrosis (IPF) offering a new promising window for both physicians and patients suffering from IPF.(2–4) The marketed formulation of PF drug product is oral hard capsule containing 267 mg of PF administered three capsules three times a day; for a total of 2403 mg/day.(5) PF is absorbed slowly and is predominantly (80%–85%) excreted via urine.
Although PF is generally well tolerated, the clinical trial studies demonstrated that a minority of patients discontinue therapy due to several adverse effects of PF after oral administration such as dyspepsia, headache, diarrhea, nausea, vomiting, fever, hepatic dysfunction dizziness, skin-related adverse effects, and facial paralysis.(4,6–8) Besides, PF has been reported to undergoes hepatic first pass metabolism by cytochrome p450 isoforms of CYP1A2, 2C9, 2C19, 2D6, and 2E1.(9) Taking together inhalation or intrapulmonary delivery of PF appears to be a good alternative to optimize drug delivery and minimize the dosage, adverse and non-specific effects.
Paraquat (PQ) is considered as a potent fibrogenic agent, which is frequently applied to produce an experimental model to study the mechanisms involved in the progression of lung fibrosis.(10–13) Lung injury caused by PQ shows lesions similar to those observed in most cases of IPF diseases in human.(13)
Therefore, the objective of this study was to recommend a novel PF-delivery route and compare it with conventional oral administration route in PQ-induced lung fibrosis rat model. This comparison was performed by evaluating morphological changes, hydroxyproline content, tissue oxidative stress parameters, proinflammatory and fibrotic genes expression including transforming growth factor beta 1 (TGF-β1), tumor necrosis factor alpha (TNF-α), tissue inhibitor of metalloproteinase 1 (TIMP-1), and matrix metalloproteinase 2 (MMP-2) genes in the lung tissues.
Materials and Methods
Materials
PF, PQ, hydroxyproline, 4-dimethylaminobenzaldehyde, chloramine T, and malondialdehyde (MDA) were purchased from Sigma-Aldrich Chemical Co. (USA). TRIzol® RNA isolation reagent and HyperScript™ first strand complementary DNA (cDNA) synthesis kit were purchased from Invitrogen (Germany). SYBR Green Master Mix was obtained from Takara (Japan). All other chemicals were bought from Merck (Germany).
Animals and treatments
Male Sprague-Dawley rats weighing 220–260 g were housed in normal laboratory conditions at 21 ± 2°C under a 12/12 light–dark cycle. Pulmonary fibrosis was induced by a single dose intraperitoneal (i.p.) administration of PQ (20 mg/kg).(11) Two weeks after the induction of fibrosis, animals were randomly divided into four experimental groups with eight animals in each group: PQ received untreated rats (PQ group), oral PF treatment [200 mg/(kg·d)], inhalation PF treatment [20 mg/(kg·d)], and negative control that received normal saline. PF in oral route was dissolved in saline and administered for 14 consecutive days by gavage. In inhalation route, PF was dissolved in saline at a concentration of 2 mg/mL and neutralized to pH 7.0.
Rats (8/chamber) were exposed to 20 mL of PF solution (∼60 min till the entire solution was evaporated) once a day for 14 consecutive days using an ultrasonic nebulizer. The volume of the chamber was about 45 L maintained under normoxic and normocapnic conditions. Selection of the dose of PF was based on the results of Poulin et al.,(14) which utilized small respirable delivered dose of 2.5 mg of PF in bleomycin-induced fibrotic lung in rats. We used twofold dose of mentioned study to ensure the efficacy of nebulized formulation. All animals were treated humanely according to the guidelines on ethics standard for investigation of experimental pain in animals and approved by the Animal Experimentation Ethics Committee of Kerman Neuroscience Research Center (EC/KNRC/90).
Sample collection and analytical procedures
At the end of the treatment period (28 days), the rats were anesthetized by injecting i.p. ketamine and xylazine (100 and 10 mg/kg, respectively). Lungs were promptly removed and divided into two halves. The left lung was immersed in 10% buffered formalin for the histopathology examination. The Right lung was stored at −80°C for the analysis of oxidative stress, hydroxyproline content, and gene expression.
Histopathology analysis
After embedding the fixed lung tissues into liquid paraffin, 5 μm thick tissue sections were prepared. Three sections for each lung were stained by hematoxylin and eosin (H&E) and Masson's trichrome staining methods. The presence and grading of inflammation(15) and fibrosis(16) were evaluated in the stained sections based on the scoring indicated in previous studies and also our previous studies.(17)
Determination of oxidative stress parameters
Preliminary preparation of lung tissue samples for biochemical evaluation was done with 0.1 M Tris-HCl buffer (pH 7.4) at 4°C using a tissue homogenizer. The resulting tissue homogenates were used for biochemical analysis. The lipid peroxidation level was measured according to the method of Ohkawa et al.(18) The MDA in the lung tissue was measured using thiobarbituric acid at 532 nm. Different concentrations of tetra butyl ammonium prepared as a standard solution. The activity of superoxide dismutase (SOD) was measured according to the method of Beyer and Fridovich(19) and catalase (CAT) activity was determined using the method of Aebi.(20)
Measurement of collagen
Hydroxyproline, as a measure of collagen deposit in the lung tissue and fibrosis, was assessed using method of Reddy and Enwemeka(21) with minor modification.(22) Briefly, hydroxyproline in the lung tissue homogenate was hydrolyzed with 1 M acetate buffer and then was oxidized with 1.4% chloramine T. A reddish purple complex with 1 M Ehrlich's reagent (4-dimethylaminobenzaldehyde) was formed and the chromophore was developed at 65°C for 20 min and measured spectrophotometrically at 550 nm.
Determination of fibrotic genes expression by real-time RT-PCR
Total RNA was extracted from pulmonary tissues using TRIzol reagent according to the manufacturer's protocol. Samples (2 μg RNA) were reverse-transcribed using a HyperScript first strand cDNA synthesis kit. Synthesized cDNA was used in real-time RT-PCR (LightCycler® 96 Roche, Germany) experiments using SYBR GREEN Supermix and analyzed with LightCycler 96 Software.
The sequences of primes were as follows(23): rat TGF-β1 (5′-GTAACGCCAGGAATTGT-3′ and 5′-CGCCATCTATGAGAAAACC-3′), rat TNF-α (5′-ACGTAGTCGGGGCAGCCTTGT-3′ and 5′-CCCACGTCGTAGCAAACCACCAA-3′), rat MMP-2 (5′-TTCAGGTAATAAGCACCCTTGAA-3′ and TAACCTGGATGCCGTCGT-3′), rat TIMP-1 (5′-GCCTCTGGCATCCTCTTG-3′ and 5′-TGCGGTTCTGGGACTTGT-3′), and rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5′-TGCTGAGTATGTCGTGGAGT-3′ and 5′-GTCTTCTGAGTGGCAGTGAT-3′). Specificity was confirmed by electrophoretic analysis of the reaction products and by inclusion of template- or reverse transcriptase-free controls. To normalize the amount of total RNA present in each reaction, GAPDH cDNA was used as an internal standard.
Statistical analysis
The quantitative data were presented as mean ± standard error of the mean. Differences between the means were analyzed by one-way analysis of variance followed with Tukey's HSD (honest significant difference) post hoc test using version 18.0 SPSS software. The differences are considered statistically significant when p < 0.05.
Results
Influence of oral and inhalation PF on lung tissue histopathology changes
Histopathology of lung tissues was evaluated by H&E or Masson's trichrome staining method. Exposure to PQ induced a marked inflammatory response characterized by inflammatory cell (lymphocytes and histiocytes) aggregation and infiltration in the alveolar space and septum, substantially thickening and degrading normal alveolar structure. Masson's trichrome stain revealed a marked increase in collagen deposition, predominately in the thickened alveolar regions and small bronchioles (Fig. 1). Inhalation and oral PF treatment attenuated the marked interstitial thickening, inflammatory responses, and collagen accumulation induced by PQ. However, it appears inhalation administration alleviated the inflammatory and fibrotic lesions more than that by oral route. All the lesions are depicted in Figure 1 and Table 1.

(To view this figure in color, please see article online at: www.liebertonline.com) Photographs of a lung specimen in rats. PQ-treated lung showing collagen fiber deposition in the thick inter-alveolar septa(s) and around pulmonary bronchioles and blood vessels. PF treated lung either in oral or inhaled route showing minimal collagen fibers in inter alveolar septa and around pulmonary blood vessels. (Magnification × 40). PF, pirfenidone; PQ, paraquat. Color images available online at www.liebertpub.com/jamp
Rats were treated with oral and inhaled PF for 14 days after development of fibrosis.
PF, pirfenidone; PQ, paraquat.
Influence of oral and inhalation PF on the oxidative stress parameters
The levels of lipid peroxidation and the antioxidant activities of SOD and CAT in the lung tissue of the control and experimental rats are depicted in Figure 2. A significant rise in the levels of lipid peroxidation in the lung tissue was observed in the PQ treated animals. This effect was accompanied by a decrease in the enzymatic activities of SOD and CAT. The administration of both oral and inhaled PF significantly decreased lipid peroxidation in lung samples (p < 0.001) and restored the SOD and CAT activity to near normal.
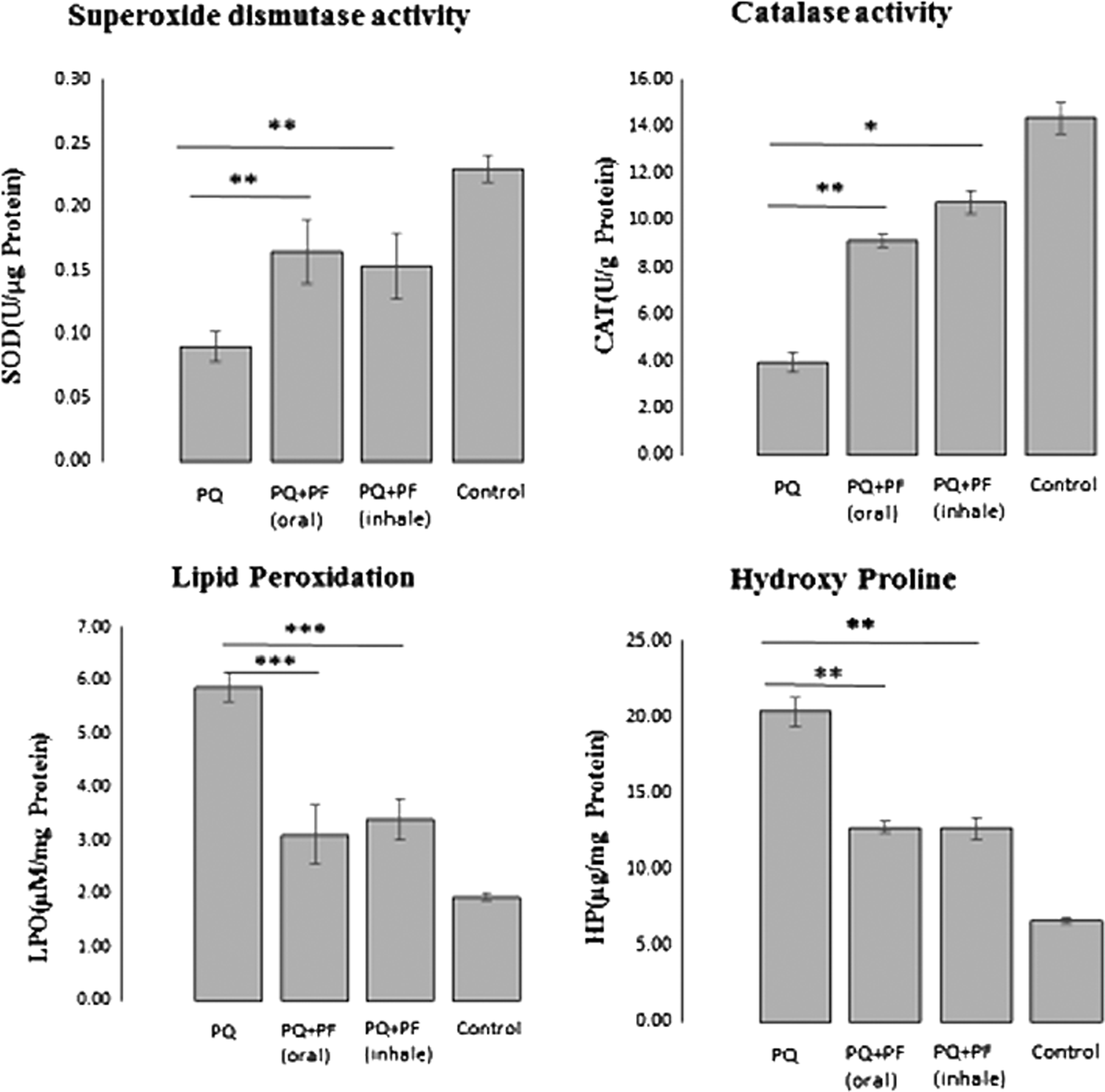
Influence of oral and inhaled PF on oxidative stress parameters and HP content. Data are expressed as the mean ± standard error of the mean (two replicates in each assay), n = 8. *p < 0.05, **p < 0.01 and ***p < 0.001 in comparison with the PQ group. CAT, catalase; HP, hydroxyproline; LPO, lipid peroxidation; SOD, superoxide dismutase.
Influence of oral and inhaled PF on hydroxyproline content of lung tissue
Hydroxyproline is the main component of collagen protein in the body. It was measured as a marker of fibrosis in the lung tissue. As shown in Figure 2, the hydroxyproline content in lungs of the PQ-treated rats significantly increased compared with the control group. Administration of PF in both administration routes significantly reduced the content of hydroxyproline in lung tissues (p < 0.01). These findings were consistent with the histological results.
Effect of oral and inhalation PF on the expression of TGF-β1 and TNF-α
To further investigate the specific antifibrotic effect of inhaled PF and comparing to oral conventional route, the expression pattern of proinflammatory and fibrosis related cytokines in lung tissues was evaluated by RT-PCR. TGF-β1 as the most important profibrotic cytokine has been demonstrated to be the most potent and direct stimulator of fibroblast proliferation and collagen production, and is requisite for pulmonary fibrosis of various etiologies.(24) As shown in Figure 3, PQ exposure progressively increased TGF-β1 while PF treatment significantly in oral and inhaled route decreased lung TGF-β1 messenger RNA (mRNA) levels. We also analyzed TNF-α that plays an important role in PQ-induced pulmonary inflammation. As expected, PQ increased lung TNF-α expression, which was significantly inhibited by both oral and inhaled PF (Fig. 3).
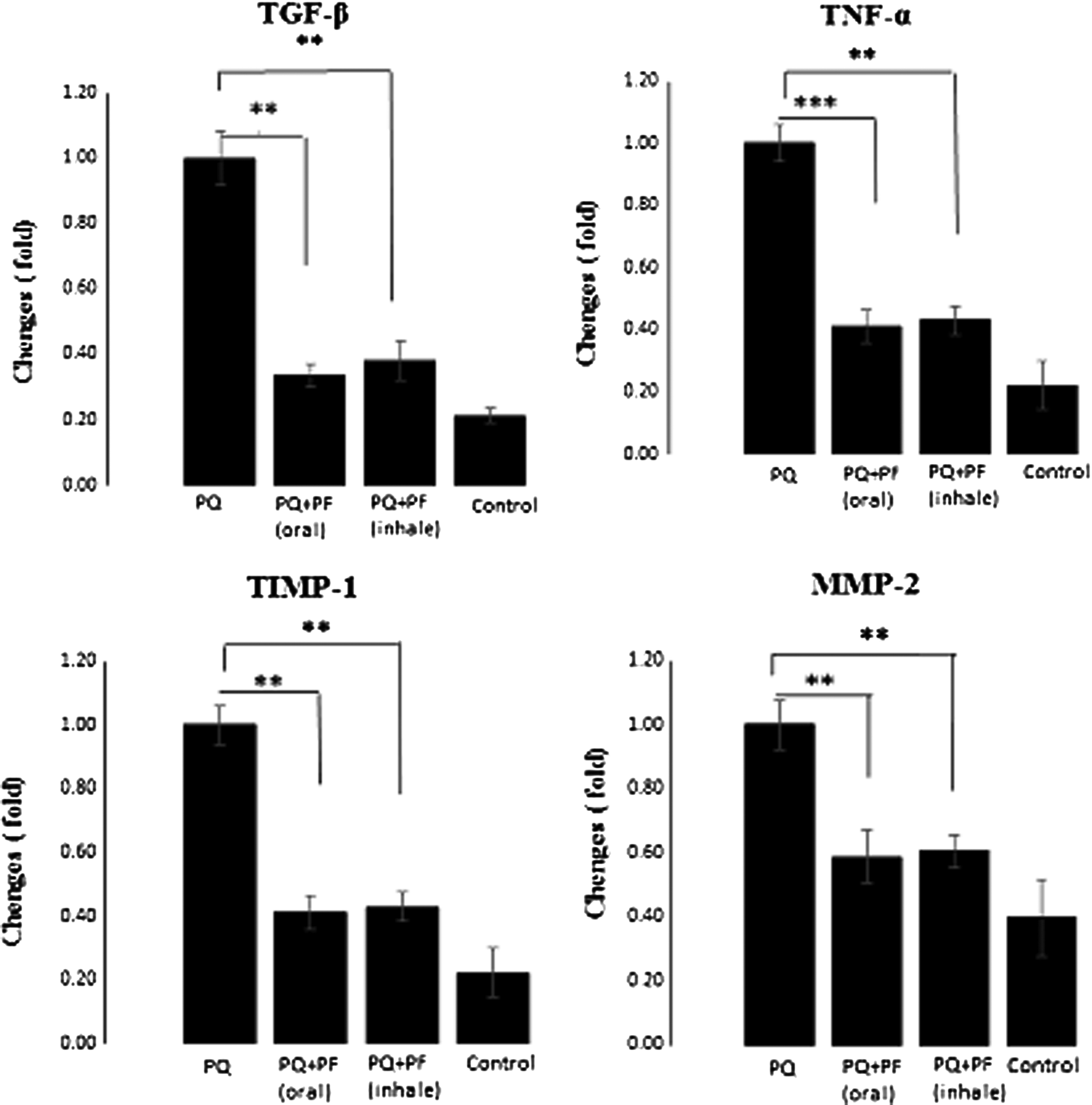
The effect of oral and inhaled PF on the expression of fibrotic genes in PQ-induced lung fibrosis. The rats treated for 14 days after development of fibrosis. The expressions of TGF-β, TNF-α, TIMP-1, and MMP-2 mRNA were determined in the lung of the rats by real-time RT-PCR. The data are means ± standard error (two replicates in each assay) for eight rats. ***p < 0.001 and **p < 0.01 in comparison with the PQ group. MMP, matrix metalloproteinase; mRNA, messenger RNA; RT, reverse transcription; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor.
Effect of oral and inhalation PF on the expression of TIMP-1 and MMP-2
Following a fibrogenic stimulus, lung cells lose their morphology and function and produce large amounts of extracellular matrix (ECM) proteins due to the overexpression of MMPs and their physiologic inhibitors TIMPs, which are proved in fibrotic lung disease. Therefore, we detected the mRNA levels of key fibrotic marker genes, TIMP-1 and MMP-2. The mRNA expression of TIMP-1 and MMP-2 genes were increased in the PQ group, however, this expression was significantly decreased in both inhaled and oral treatment, which suggested that PF in both inhaled and oral treatment groups could inhibit the activation of ECM deposition and the progression of fibrosis (Fig. 3).
Discussion
Besides histopathology examination, the expression pattern of inflammatory and fibrosis-related genes along with hydroxyproline content and antioxidant status in the lung tissues was evaluated to elucidate the underlying mechanisms responsible for the observed effects of PF, administered both via inhalation and oral routes. In comparison to the other organs, lungs are exposed to the highest amount of oxygen and, therefore, more subsequent oxidative damage.(25) As always denoted, fibrosis originally results from an imbalance between reactive oxygen species (ROS) generation and antioxidant defenses. Oxygen free radicals cause the oxidation and damage of genetic materials, proteins and membranous lipids. It has been demonstrated that oxidative stress inhibitors provide beneficial effects in lung fibrosis.(25)
In this study, a significant rise in the levels of lipid peroxidation in the lung tissues was observed in the PQ-treated animals. This effect was accompanied by a decrease in the enzymatic activities of SOD and CAT. Administration of inhalation PF was comparable to oral PF in decreasing lipid peroxidation and restoration of SOD and CAT activities close to normal, indicating reduced ROS formation may play a role by which the antifibrotic effects of the PF administered via both routes are mediated. Consistent with lipid peroxidation and antioxidant findings, histological evaluation in lung tissue indicates a marked alleviation in both oral and inhalation PF groups, which confirms the antifibrotic effects of PF. Interestingly, administrated PF by inhalation appears to be a drug delivery to reduce dosage and nonspecific effects and direct drug delivery to the lungs.
In addition, administration of PF via inhalation inhibited the hydroxyproline content comparatively greater than that by oral route. This may indicate attenuation of PQ-induced lung injury by PF particularly after its administration via inhalation.
Recently, there has been a growing understanding of the fibrosis pathophysiology, and it has been shown that PQ applies as a potent fibrogenic agent to study the mechanisms involved in the progression of lung fibrosis and induces lesions similar to those observed in IPF disease in human.(13,26)
In this study, it is shown that PQ-induced increase in expression of TGF-β was significantly reversed by inhalation and oral PF treatment. The role of TGF-β1 in the progression of fibrosis in different organs is well documented and inhibition of this pathway has been introduced as one of the suitable approaches to prevent fibrosis development.(27–29) This suggests that both routes of PF administration exert their antifibrotic effects partly through inhibition of TGF-β1 signaling.
Conte et al.,(30) have shown that PF modulates primary human lung fibroblasts proliferation and TGF-β-mediated differentiation into myofibroblasts by blocking the TGF-β1-induced signaling pathways. Nakayama et al.,(31) have reported the inhibitory effect of PF on the expression of HSP47 in TGF-β1-stimulated human lung fibroblasts. Choi et al.,(32) have stated that PF inhibits TGF-β1-induced fibrogenesis by blocking nuclear translocation of Smads in human retinal pigment epithelial cell line ARPE-19. Hisatomi et al.,(33) have noticed that PF inhibits TGF-β1-induced overexpression of collagen type I and heat shock protein 47 in A549 cells.
In addition, it has been denoted that some proinflammatory and profibrogenic cytokines such as TNF-α and TGF-β1 play an important role in pulmonary inflammation and fibrosis.(12,33) The interactive effect between oxidative stress and TGF-β is also reported in the progress of fibrosis.(25,34) TGF-β1, the most important mediator of fibrogenesis, plays a vital role in differentiation of fibroblasts into active myofibroblasts and inhibition of fibroblast apoptosis, epithelial-mesenchymal transition, and production and deposition of collagen and ECM.(24,35,36) TGF-β1 stimulates ROS production and oxidative stress while oxidative stress activates latent TGF-β1, a vicious profibrogenic circle. As over fibro-proliferation and ECM deposition are critical determinants of fibrogenesis, TGF-β inhibition is an appropriate marker for depiction of reduced fibrosis development in experimental models.(34)
PQ-induced ROS can also activate intracellular transcription factors such as nuclear factor (NF)-κB, thereby enhancing TNF-α. TNF-α is one of the early cytokines released from alveolar macrophages in the early stages of inflammatory phase and late stages of fibrotic phases in lung injury(33) and consistently found in animal models of pulmonary fibrosis. TNF-α activates NF-B and activator protein-1 that regulate the proliferation of fibroblasts and expression of cellular matrix deposition genes, such as MMPs and the release of some proinflammatory cytokines. The overexpression of MMPs and their physiologic inhibitors, TIMPs, are proved in fibrotic lung diseases.(37,38) MMPs are a family of ECM degrading enzymes secreted from inflammatory cells having critical roles in inflammation, tissue remodeling, and development of fibrosis.(38) Interestingly, TGF-β1-induced lung fibrosis is found primarily due to TIMP upregulation,(39) and it is reported that MMP-2 is widely expressed in fibrotic lungs.(37) Furthermore, if TIMPs are elevated in fibrosis, any agent that could reduce the secretion of TIMP by fibroblasts is possibly a powerful tool in the therapy of this devastating disease.(39)
In this study, significant increase in MMP-2 and TIMP-1 were observed in rats subjected to PQ exposure. Increased MMP-2 expression preceded an elevated TIMP-1 expression; a pattern that supports imbalance between matrix degradation and its inhibition may contribute to PQ-induced pulmonary fibrosis. PF in this regard, either given orally or via inhalation, resulted in decreased MMP-2 and TIMP-1 levels indicating correction of PQ-induced overexpressed MMP-2/TIMP-1 ratio.
Although the differences between PF impacts in oral and inhalation routes were not notable, however, the differences between administered doses were around 10-folds. This may suggest that inhalation PF may act more efficient and more specific. The present study first provided the evidence that inhalation administration of PF can be used as a drug delivery with a much lower dose needed and a higher specificity. Second, the data presented here supports the involvement of several responsible mechanisms underlying PF-mediated antifibrotic effect; that is, it may involve (1) inhibition of TGF-β1 expression; (2) inhibition of TNF-α expression; and (3) correction of the PQ-induced imbalance of MMP-2/TIMP-1 ratio.
The current study also had several limitations. For example, considering the wide range of biologic effects of PF, there may be far more complex mechanisms than we had mentioned in this article. Larger animal studies and clinical investigations are necessary to further define the benefit of PF inhalation administration route. In addition, therapeutic mechanisms of PF in lung injury treatment should also be both dose-dependently and time-dependently assessed.
Conclusion
In conclusion, PF offers great potential as an inhalation drug delivery for treating pulmonary fibrosis by modulating expression pattern of inflammatory and fibrosis-related genes in addition to hydroxyproline content and antioxidant status in the lung tissues. Therefore, this route of administration would be a particularly useful alternative to optimize drug delivery and minimize the dosage, adverse and nonspecific effects.
Footnotes
Acknowledgments
The authors acknowledge Prof. Mahmoud Ghazi-Khansari, the Department of Pharmacology, School of Medicine, Tehran University of Medical sciences for providing Paraquat poison and advice aiding successful completion of this study. This work was supported by the Iran National Science Foundation (INSF) (No. 93021576).
Author Disclosure Statement
All authors declare that no conflict of interest exists.