
Abstract
Abstract
Background:
As non-cystic fibrosis bronchiectasis (NCFB) progresses, patients suffer irreversible lung damage and deterioration in lung function. This study explored whether inhalational parameters (peak inspiratory flow [PIF, primary endpoint], inspiratory volume and time [secondary endpoints]) represent barriers to complete dosing in patients with poor lung function who are using Ciprofloxacin dry powder for inhalation (DPI) (a drug–device combination of the T-326 inhaler device and a Ciprofloxacin dry powder formulation).
Methods:
This open-label, multicenter study generated inspiratory flow rate data from patients with NCFB using the breath-actuated T-326 dry powder inhaler. These rates were compared against reference values to identify whether patients with all degrees of lung function impairment could generate sufficient flow rates to facilitate adequate drug delivery. Patients attended screening and a second visit 1 − 14 days later. Forced expiratory volume in one second (FEV1), forced vital capacity (FVC), FEV1/FVC, and inspiratory capacity were measured via spirometry at both visits. Forty-two patients were screened for inclusion; 33 met eligibility criteria and were stratified into one of three groups based on their FEV1% predicted value (group 1: 25% ≤ FEV1% predicted <45%; group 2: 45% ≤ FEV1% predicted <70%; group 3: FEV1% predicted ≥70%).
Results:
No significant between-group differences occurred in PIF (mean flow rates 68.21, 66.01, and 65.18 L/min in groups 1, 2, and 3, respectively). Individual minimum PIFs of 46.0–49.0 L/min were observed across groups. These results all exceeded the reference value (minimum PIF 45 L/min for Ciprofloxacin DPI) indicating that regardless of the level of airflow obstruction, patients were capable of achieving sufficient PIFs to aerosolize and inhale Ciprofloxacin dry powder with the T-326 inhaler.
Conclusions:
Our data indicate that T-326 is suitable for use in the drug–device combination Ciprofloxacin DPI to provide targeted pulmonary delivery in patients with NCFB, including those with significantly impaired lung function.
Introduction
Inhalation therapies for lung diseases offer key benefits in the form of targeted drug delivery and reduced systemic exposure. Delivery of the drug directly to the lung can be facilitated by several drug–device systems, including nebulizers, pressurized metered-dose inhalers, and dry powder inhalers,(1) as well as by the use of particular drug formulations.
Optimal inhaled therapy requires a fine balance between the characteristics of both drug and device on one hand, and the patient's lung function on the other.(1,2) The flow rates that are generated through a device determine dispersion of a dry powder and depend on both device resistance and lung function,(3) meaning that a patient's ability to inhale drugs in an efficient manner may be reduced when there is significant impairment of lung function.
Previous research has shown that reductions in peak inspiratory flow (PIF)(4,5) and inspiratory volume (IV)(4) reduce the lung deposition of dry powder for inhalation (DPI). These parameters are therefore of paramount importance when considering the suitability of a combination of an inhaler and a DPI formulation for patients with impaired lung function.(5) Data pertaining to the relationship between PIF, forced expiratory volume in one second (FEV1), and inspiratory capacity (IC) in patients with non-cystic fibrosis bronchiectasis (NCFB) are lacking.
Dry powder inhalers vary considerably with respect to the characteristics of the ideal inhalation maneuver required to achieve effective drug delivery.(6) The T-326 inhaler (Novartis, San Carlos, CA; Fig. 1) is a hand-held, breath-actuated inhaler for use with hydroxypropylmethyl cellulose capsules containing a dry powder drug formulation. In vitro studies have demonstrated that a minimum flow rate of 45 L/min and a minimum volume drawn through the T-326 inhaler of 1.3 L are sufficient for complete aerosolization and inhalation of a 50-mg clinical dose of Ciprofloxacin dry powder.(7) The powder itself is optimized for pulmonary deposition. It has a mass median aerodynamic diameter of ∼3.6 μm with a geometric standard deviation of 2.1. This results in a fine particle fraction (<4.5 μm) of 51.5%(7) and a mean (SD, range) intrapulmonary deposition of 53% (11%, 38–64%) independent of respiratory function.(8)
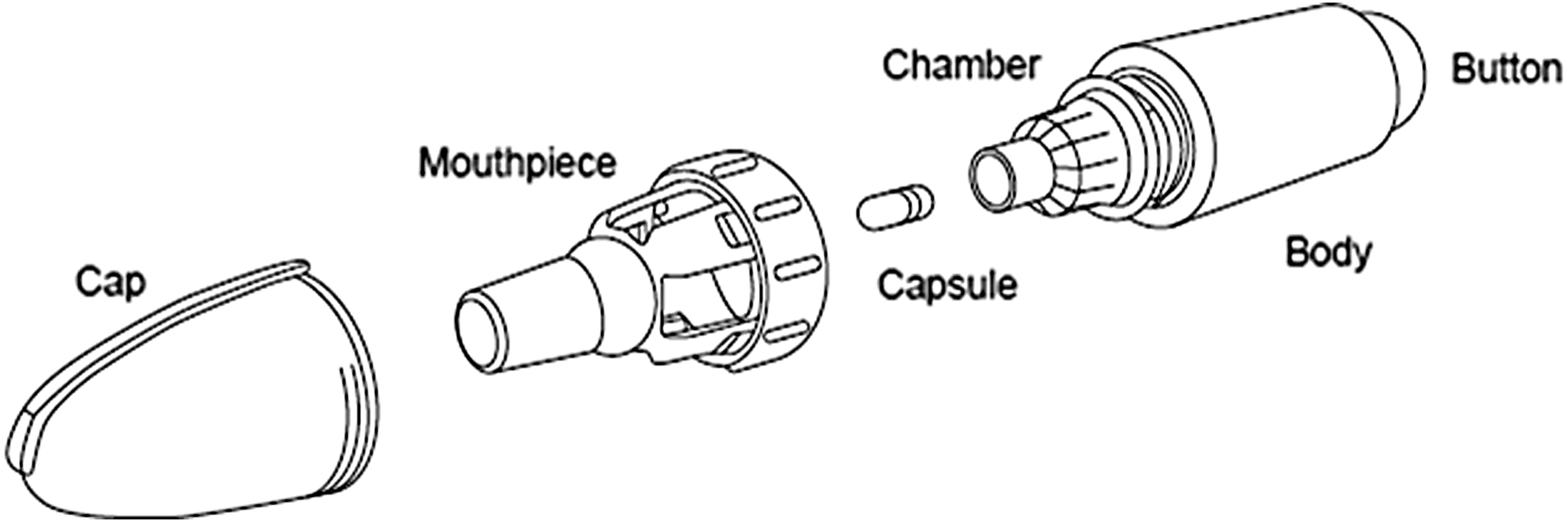
Structure of the T-326 inhaler.
The aim of the current study was to measure inhalation flow rates in patients with NCFB using the T-326 inhaler to determine whether specific patient-related parameters (PIF, primary endpoint; IV and time, secondary endpoints) present a barrier to complete dosing for patients with poor lung function. These data will help to determine whether this specific device is suitable for combination in a dry powder–device combination for use in clinical practice irrespective of patients' lung function.
Materials and Methods
Study design
This phase I study followed an open-label, multicenter (N = 9) design and was conducted in Germany between November 23, 2015, and February 18, 2016. The study was sponsored by Bayer AG, Germany.
Patients attended a screening visit (visit 1) followed by a second visit 1 − 14 days later (visit 2), at which point flow profiles were measured and patients were stratified into three groups based on FEV1% predicted. Spirometry was used to measure the following lung function parameters at both visits: FEV1, forced vital capacity (FVC), FEV1/FVC, IC (the maximum volume of air that can be inspired after normal exhalation), and IV (the maximum volume of air that can be inspired when using the inhaler device). A telephonic follow-up was conducted 1 − 7 days after visit 2 with the aim of evaluating patient safety. The total study duration for each patient was between 3 and 22 days.
The intention was that the study would recruit sufficient patients to provide at least 10 evaluable datasets in each of the three groups. Further information on patient stratification is given below. The conduct of this clinical study met all local legal and regulatory requirements, and was compliant with the ethical principles enshrined in the Declaration of Helsinki; the International Conference on Harmonization guideline E6: Good Clinical Practice; and the specification for medical devices—DIN EN ISO 14155. Written informed consent was obtained from each patient before participation in the study.
Patients
NCFB patients with varying degrees of lung function impairment were recruited from nine pulmonology practices in Germany. To be eligible for inclusion in the study, patients had to be 18–85 years old, with a documented diagnosis of NCFB (made using either computed tomography scan or magnetic resonance tomography) involving two or more lobes, and dilated airways compatible with bronchiectasis at the time of initial diagnosis. Patients were required to have stable disease together with the ability to perform spirometry reproducibly in line with the criteria issued by the American Thoracic Society/European Respiratory Society.(9) Patients were able to continue to take any currently prescribed bronchodilators or other inhaled therapies during the course of this study.
Patients were ineligible for the study if they met any of the following criteria: presence of a pulmonary exacerbation or hemoptysis within 6 weeks of the study visit; an FEV1 < 25% of the predicted value for the age, height, and gender of the patient; any history or evidence of lung transplant, lung resection, thoracotomy, known aneurysm, or severe scoliosis; an established diagnosis of bronchial asthma or cystic fibrosis; current pregnancy or breastfeeding; and exposure to any investigational agent or involvement in another medical device study within the 4 weeks preceding screening or at any point during the study.
Patient stratification
At the second visit, patients were stratified into three groups based on their FEV1% predicted value for age, height, and gender according to the European Coal and Steel Community (ECSC) criteria.(10) The groups were defined as follows: group 1: 25% ≤ FEV1% predicted <45%; group 2: 45% ≤ FEV1% predicted <70%; group 3: FEV1% predicted ≥70%.
Patient withdrawal from the study
Withdrawal from the study was mandatory in the event that it was requested by the patient or the patient's legally acceptable representative, or where there was a violation of the inclusion or exclusion criteria. Patients could also be withdrawn from the study at the request of the study sponsor, or if the investigator considered that continuation would be harmful to the patient's well-being. Reasons for patient withdrawal were recorded, and any patient removed due to an adverse event (AE) was monitored until symptoms stabilized or subsided. Wherever possible, patients recorded as dropouts were replaced until 10 evaluable datasets per group had been recorded.
Study procedures
The study comprised three stages: patient training, data collection and computation (patient inhalation maneuvers and flow profile measurement), and comparison of the data against reference values.
Patient training
All patients received instruction regarding the correct inhalation maneuver from the principal investigator or a designated member of the study team. Patients were instructed to sit or stand in an upright position and exhale one full breath. Holding the inhaler horizontally, patients were then asked to close their lips around the mouthpiece and inhale deeply and comfortably. Patients were instructed not to exhale into the device. Each patient performed a series of test inhalations, which were assessed for compliance with the instructions.
Data collection and computation
Patient inhalation maneuvers
Once the correct inhalation maneuver had been confirmed, patients provided three consecutive inhalation measurements using the T-326 inhaler. The device was loaded with an empty capsule to allow for any additional aerodynamic resistance caused by the capsule during the inhalation process. A single empty capsule was used for each inhalation. Patients were allowed at least 2 minutes of rest between maneuvers.
Measurement of flow profiles
Inhalation flow rate was assessed as a function of time by measuring patient-generated differential pressure within the mouthpiece of the inhaler against ambient pressure. Differential pressure was measured using a mouthpiece adaptor with a probe connected to a pressure transducer. The differential pressure measured at the mouthpiece was converted into a flow rate using a validated flow profile measurement system (Inamed GmbH, Gauting, Germany) based on a correlation curve representing the association between differential pressure and flow rate of the T-326 inhaler.
The correlation curve was generated before the start of data collection by measurement in parallel of the differential pressure (using the flow profile measurement system; Inamed proprietary software) and the flow rate (using a mass flow meter; Model 4040; TSI, Aachen, Germany) when applying a flow profile generated by a computerized flow/volume simulator (BRS3000; Copley Scientific, Nottingham, United Kingdom).(11) The resulting flow profiles of each patient were analyzed with respect to the study endpoints and compared against the reference values. This measurement procedure has previously been employed in flow profile studies that support the approval of generic inhalation drug products.(12,13)
Comparison of data with reference values
To evaluate the clinical implications of the results, patient data were compared against reference values determined by cascade impactor and laser diffractometric measurements for the minimum PIF (45 L/min) and minimum IV (1.3 L) required to empty the contents of a capsule containing 50 mg of Ciprofloxacin DPI.(7) These cut-offs were used here to identify those patients likely to be able to achieve adequate lung deposition of a full dose.
Study endpoints
The primary outcome variable in this study was PIF (L/min) achieved with the T-326 inhaler. The secondary outcome variables were IV (V) in liters and inspiratory time (t) in seconds.
Safety endpoints included all clinically significant findings from physical examination, vital signs, pulse oximetric measurements, lung function measurements by spirometry, AEs, and adverse device effects. All AEs recorded during the study were assessed for seriousness, intensity, and causal relationship with the device/drug.
Populations for analysis
The following patient populations were considered for statistical analysis. The safety analysis (SAF) population consisted of all patients who had completed at least one inhalation through the study device. The per protocol (PP) population consisted of all patients from the SAF population for whom technically acceptable flow profiles were recorded at visit 2 as specified in the clinical study protocol, and who had no major protocol deviations. Screening failures were captured in the study database, but no further analyses were performed on this subgroup.
Statistical methods
Descriptive statistics were applied to the primary and secondary study endpoints. Statistical analyses were performed on the data obtained from all patients stratified according to FEV1% predicted values. Missing data were not substituted, and all patients who dropped out of the study were included within the appropriate analysis population. Of the three evaluable inspiratory maneuvers, the maneuver with the maximum PIF rate for each patient was selected for inclusion in the descriptive analyses.
Results
Patients
A total of 42 patients were screened for inclusion in this study. Of these, nine constituted screening failures, while the remaining 33 patients met the eligibility criteria and were assigned to a group. All 33 eligible patients completed the study (Fig. 2), meaning that the SAF and PP populations were identical. Examination of patient characteristics revealed that group 1 contained proportionally more men than the other two groups, and had a lower mean age (48.7 vs. 63.1 and 61.3 years for groups 2 and 3, respectively) (Table 1).
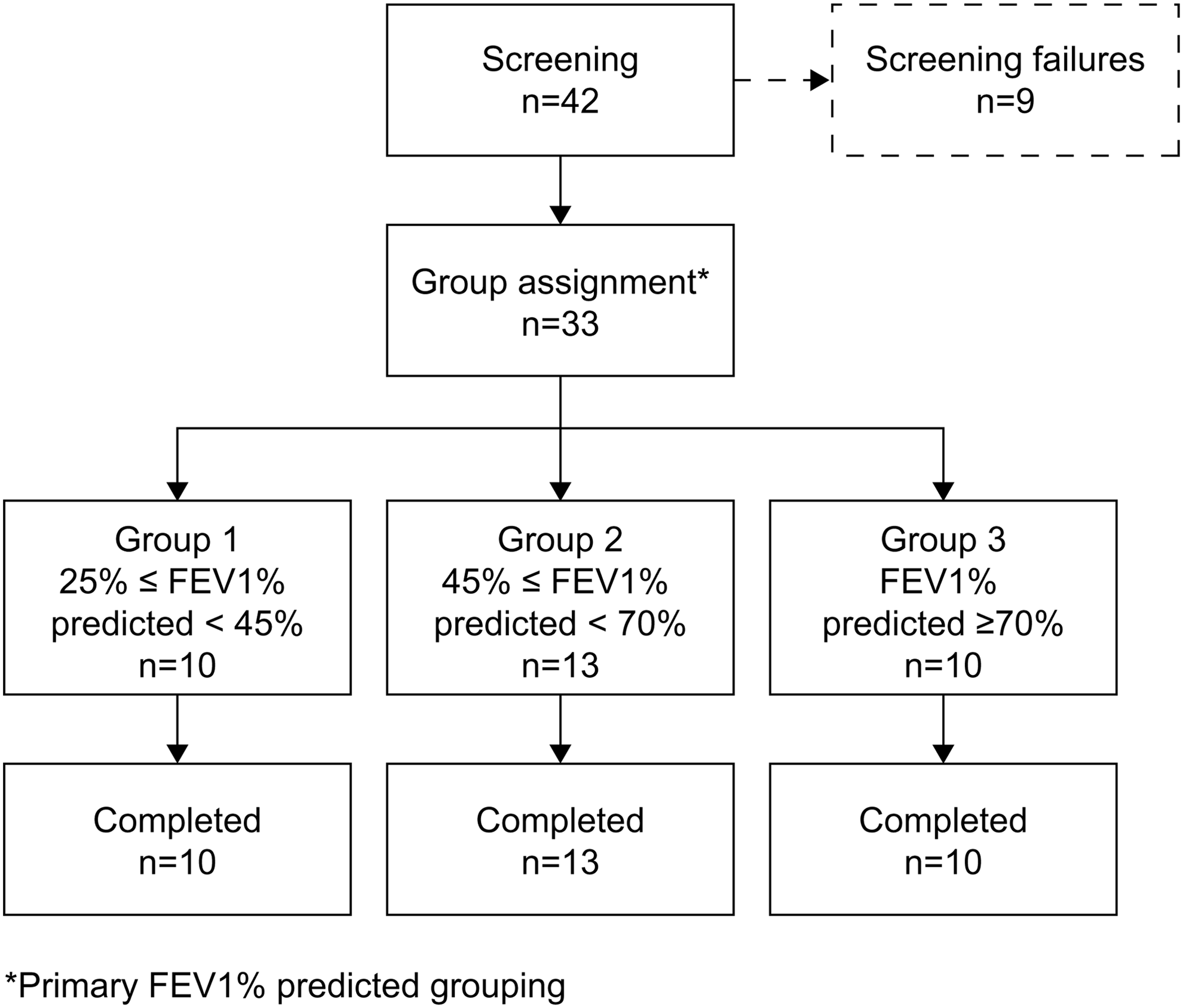
Patient disposition throughout the study by primary FEV1% predicted grouping. FEV1, forced expiratory volume in one second.
Baseline Demographics and Patient Characteristics (SAF Population)
FEV1, forced expiratory volume in one second; FVC, forced vital capacity; IC, inspiratory capacity; SAF, safety analysis.
Across the whole sample, FEV1 values ranged from 26% to 121% of that predicted on the basis of the patient's age, height, and gender. It should be noted, however, that the spirometry results indicate that FEV1% predicted was a poor predictor of IC, with higher mean IC values observed among patients in group 1 (mean IC 1.9 L) compared with those in group 2 (mean IC 1.6 L) (Table 1 and Fig. 3), perhaps as a consequence of the greater proportion of men within group 1.

Box plot of IC (L) (SAF population/PP population) stratified according to primary FEV1% predicted patient grouping. IC, inspiratory capacity; PP, per protocol; SAF, safety analysis.
Primary endpoint
The primary outcome variable for this study was PIF (L/min) as measured at visit 2 (Fig. 4a). Inspiratory flow was comparable between all three groups, with means of 68.2, 66.0, and 65.2 L/min being recorded in groups 1, 2, and 3, respectively, and individual minimum PIFs ranging between 46.0 and 49.0 L/min. Patients with both low and high IC were equally capable of achieving adequate PIFs (Fig. 4b).

Secondary endpoints
Those patients with the lowest FEV1% predicted (group 1) demonstrated IV values that were comparable to those achieved by patients with the highest FEV1% predicted (group 3). Mean IVs were 2.6, 1.9, and 2.6 L for groups 1, 2, and 3, respectively, with individual minimum IV values ranging from 0.6 to 3.3 L (Fig. 5a). According to the reference values, an IV of at least 1.3 L is required to ensure that the capsule is fully emptied in one inhalation.(7) Thirty-one of 33 patients (93.9%) achieved an IV above this threshold in a single inhalation maneuver. As expected, IVs were positively correlated with inspiratory capacities: patients who demonstrated an IV <1.3 L also demonstrated the lowest inspiratory capacities (Fig. 5b).

Mean inspiratory times were 3.1, 2.4, and 3.6 seconds for groups 1, 2, and 3, respectively, and were comparable between patients with high and low FEV1% predicted. Minimum values ranged from 0.9 to 2.4 seconds across the three patient groups.
Safety
The 33 patients who had performed at least one inhalation using the T-326 inhaler were included in the SAF population for analysis. All 33 successfully completed three inhalation maneuvers at visit 2. One patient in group 2 experienced a treatment-emergent AE after using the T-326 inhaler. This was a short episode of mild coughing that did not require medical intervention, and which was considered by the investigator to be procedure-related rather than device-related. No deaths, serious AEs, or other significant AEs occurred during this study.
Discussion
This study sought to measure the inspiratory flow achieved with the T-326 inhaler in patients with NCFB who present with differing degrees of lung function impairment. Adequate flow rates must be generated to provide the energy for effective powder dispersion,(3) and the T-326 inhaler has been designed to ensure a low airflow resistance.(14)
The findings from this study demonstrate that the aerodynamic characteristics of the T-326 inhaler are suited for effective drug delivery in a heterogeneous population of patients with NCFB and impaired lung function. All 33 patients with NCFB, including those with the lowest FEV1% predicted, attained a PIF >45 L/min, which is the minimum rate required for effective capsule-emptying using the device.
Patients were capable of achieving PIFs ≥45 L/min independently of their IC, as shown in Figure 4b. This indicates that even when there is severe airflow impairment, patients can generate a sufficient flow rate with the T-326 device to inhale a complete dose. The findings support earlier unpublished data that suggest that both PIF and IV thresholds must be met for effective dose delivery. In those patients with an IV <1.3 L, two inhalation maneuvers may be required to ensure that complete delivery of the dose is achieved. These findings emphasize the importance of conducting PIF studies in target populations to validate in vitro laboratory testing as an integral part of the clinical development plan, even in cases where a commercially available device is used.
The T-326 inhaler had a favorable safety profile, with only one patient reporting a treatment-emergent AE (short episode of mild coughing), which was not considered to be device-related. It should be noted that the breath-actuated nature of this inhaler relies on effective patient education to ensure successful and reproducible lung deposition of the therapeutic agent.
The patients enrolled in this study were stratified according to their FEV1% predicted at baseline, which at present continues to be the most widely used diagnostic parameter to assess patient lung function.(15) However, the present data revealed FEV1% predicted to be a poor predictor of patients' ability to achieve the minimum IV required for effective drug delivery in one breath from the T-326 inhaler. Therefore, patient stratification according to FEV1% predicted, despite being the current standard recommended by ECSC, does not appear to provide a meaningful assessment of a patient's ability to use the inhaler device effectively.
Effort, technique, and respiratory muscle strength are all important factors underlying an individual's ability to generate an adequate PIF to deliver an inhaled drug to the lungs. Independent predictors other than PIF include IC% predicted and FVC% predicted, and in a separate study, both inspiratory and expiratory mouth pressures were found to correlate with PIF.(5)
These observations have important implications for the translation of research findings into instructions for device use in an outpatient setting. The study data indicate that patients with poor IC rather than low FEV1% predicted (Fig. 5b) should use a second inhalation maneuver to generate a high enough IV when using the T-326 device. For all other patients, one inhalation maneuver should be sufficient to deliver the complete dose to the respiratory tract.
Although IC is related to the ability to generate a sufficient IV, it is impractical as a measure of lung function in outpatients. Therefore, rather than stratifying patients according to clinical parameters, a more conservative rule could be applied—for example, that all patients should perform two inhalation maneuvers regardless of the severity of their lung function impairment. This is consistent with the advice given for other capsule-based devices.(3,16)
Limitations of this study include the fact that minor disparities were seen in the demographic characteristics within and across the different groups. Notably, the patients recruited to group 1 were younger than those recruited to the other study groups. Recruitment of large numbers of patients with an orphan disease is challenging and becomes more so when trying to age-match across groups defined by lung function. Although lung function as measured by FEV1% predicted is known to decline with age, the current data indicate that this should not be a primary determinant of a patient's ability to use the T-326 device.
In conclusion, the data obtained in this study suggest that the T-326 inhaler is suitable for combination with Ciprofloxacin dry powder as a drug–device combination for targeted pulmonary delivery in NCFB patients, including those with significantly impaired lung function.
Footnotes
Acknowledgments
This study was conducted by Inamed GmbH and Chrestos Concept GmbH & Co. KG, and sponsored by Bayer AG. The authors would like to acknowledge highfield:communication for the provision of medical writing and editorial services with funding from Bayer AG. Trial registry identifier: ![]() ; identifier: NCT02563197.
; identifier: NCT02563197.
Author Disclosure Statement
H.S. and J.N. are employees of Bayer AG. D.K., K.S., and A.P. are employees of Inamed GmbH. B.W. is the director of Chrestos Concept GmbH & Co. KG.
Reviewed by:
Sneha Dhapare
James Blanchard