
Abstract
Abstract
Background:
A highly potent pan-Janus kinase (JAK) inhibitor with excellent kinome selectivity was developed for topical delivery to treat severe asthma. This poorly soluble drug discovery candidate, iJAK-001, is expected to exhibit long duration of JAK/STAT pathway inhibition at low doses in asthmatics because of depot effect after dry powder inhalation. Human dose projection for inhaled molecules with low aqueous solubility remains to be a daunting challenge because of several limitations: (1) bioanalytical measurement of dissolved fraction after inhalation of solid particles is uncertain; (2) distribution of these particles is not homogenous in the lung; (3) in vitro solubility measurements to estimate fraction dissolved may not be a reflection of local surface lung concentration; (4) lack of a surrogate biomarker of lung target engagement, and (5) invasive procedure needed to sample human lung tissue in the clinic.
Methods:
We leveraged in silico, in vitro, and in vivo tools preclinically and found significant differences in lung to plasma partition ratio when iJAK-001 was given intravenously (IV) or intratracheally in a solution-based formulation versus that in suspension, as well as pharmacodynamic response in preclinical asthma models when delivered systemically via IV infusion versus inhaled.
Results and Conclusion:
The combined results from above suggest that caution must be exercised using either lung or plasma exposure for human dose projection. Instead, using the local inhibitor concentration estimate based on delivery efficiency, dose, fraction absorbed, and rate of absorption normalized by lung (cardiac) blood flow may be more appropriate for dose projection.
Introduction
Controlling severe asthmatic symptoms continues to be a medical challenge and health care burden for the general public. Chronic treatment with high doses of inhaled corticosteroids (ICS) in combination with either short or long acting β2 agonists or long acting muscarinic receptor antagonists to severe asthmatic patients offers somewhat effective therapy. However, there is still an urgent need for novel asthma therapy because of steroid insensitivity and/or unwanted side effects associated with either long-term oral or inhaled steroid use controlling the symptoms.(1–3) Oral pan-Janus kinase (JAK) inhibitors have been proposed to provide a new treatment option for such complications. For example, tofacitinib has been approved for the treatment of autoimmune-related diseases such as rheumatoid arthritis.(4,5) In addition, a recent publication suggests that pan-inhibitors may have synergistic effects with steroids, especially as the former has the potential to better modulate non-Th2-related signaling disease pathways such as INF-γ-induced asthma attack.(6,7) Despite the anti-inflammatory promises offered by this drug class, there is significant concern with oral delivery as, at high enough doses, this drug class has been associated with adverse events such as anemia, elevated low-density lipoprotein cholesterol, and increased risk of infection and malignancy.(8–10) Therefore, the approved dose for tofacitinib is currently capped at 5 mg BID (twice per day) (tablet) or 11 mg QD (once per day) (extended release). However, our PBPK (physiologically based pharmacokinetics) modeling exercise, based on in-house preclinical data,(11) published clinical PKPD (pharmacokinetics-pharmacodynamics) relationship of tofacitinib(4); we hypothesized that oral delivery of a pan-JAK inhibitor might require higher and more frequent doses compared with inhaled delivery to offer therapeutic promise for severe asthmatic patients.
Therefore, to provide novel treatment options for severe asthma, it is theorized that an inhaled pan-JAK inhibitor might offer a therapeutic advantage with lower probability of systemic side effects over oral JAK inhibitors. In the optimization process, a very potent and kinome selective JAK inhibitor, iJAK-001 (U.S. patent 20160280704), was identified with a desired solubility profile that is suitable for inhaled delivery. That is, on the one hand, the compound has low enough aqueous solubility in simulated lung fluid (SLF) so that when inhaled or dosed intratracheally (IT), as either dry powder or fine suspension, the compound exhibited a long duration of action (i.e., depot effect) similar to some of the ICS such as mometasone and fluticasone.(12) On the other hand, despite its low solubility, the fraction dissolved after inhalation was high enough so that sufficient amount of the inhibitor was released/absorbed by the epithelial lining of the lung, from either fine particle suspensions or solid dry powder, to provide consistent and robust pharmacodynamic (PD) response in diseased animal models.
Human dose projection and translational PKPD for inhaled drugs continue to be a major scientific challenge. For instance, some publications have cautioned against using total lung homogenate concentration or bronchial lavage fluid from in vivo samples as indicators of systemic target engagement(13,14) as it is not trivial to determine fraction dissolved of poorly soluble inhaled drugs. In addition, internal experience with an exploratory inhaled molecule with very low solubility showed a nonhomogenous distribution throughout different regions of lung tissues, which is consistent with literature.(15,16) Furthermore, particle size of the inhaled dry powder has been shown to affect how deep the solid particles can travel down the airway(17); for instance, particles with aerodynamic particle size of less than 2 μm have higher probability of reaching the deep alveolar sacs in the peripheral lung region, whereas larger particles (∼5 μm) are likely to be trapped in the central lung region, mainly in the trachea or upper bronchus. Finally, the amount of drug available for target engagement after dry powder inhalation is almost always less efficient than fine particle suspension delivered IT.(11) A lack of efficacy might be a reflection of poor delivery efficiency, insufficient amount of solid particles dissolved, poor distribution in the lung, or insufficient free concentration available for receptor binding after absorption. Taken together, these challenges make interpretation of preclinical PKPD data extremely challenging.
To overcome these challenges, we propose the following systematic approaches to estimate fraction dissolved in vivo after IT delivery of iJAK-001. These approaches include the following: (1) SimCYP prediction of free lung to plasma partition ratio based on experimentally measured log P, pKa, and passive permeability, (2) measurement of kinetic and thermodynamic solubility from dry powder in SLF, (3) comparison of free lung to plasma partition of iJAK-001 after IV (solution), IT (solution), and IT (suspension) dosing in rats, and (4) comparison of PD response of IJAK-001 in rat and sheep after single or multiple inhalation doses or inhalation versus IV infusion. The intention is for the outcomes of these experimental approaches to aid in human dose prediction.
Materials and Methods
Chemicals
iJAK-001 was synthesized in-house based on a general procedure published in U.S. patent 20160280704 and micronized to a particle size of D90 < 5 μm. SLF was prepared based on literature.(18)
Aqueous solubility of micronized iJAK-001 in SLF
Solubility of iJAK-001 in SLF was measured by suspending milled iJAK-001 from above with stirring in SLF at concentrations of 0.03, 0.1, 1, and 3 mg/mL at room temperature for 4 and 24 hours. At each time point, 1000 μL aliquot of the suspension was centrifuged at 16,000 g for 10 minutes at 25°C. After repeating the process twice, the supernatant was diluted 2 × by adding 100 μL of the sample to 100 μL of acetonitrile:water (1:1), and the experiments were performed in duplicate. A fit-for-purpose UPLC (ultra-performance liquid chromatography) method was developed using a Waters Acquity BEH C18 column (Waters Corp., Milford, MA) at a flow rate of 0.5 mL/min with UV detection wavelengths set at 210 and 254 nm for the solubility determination. The mobile phase consisted of isocratic mixture of 1:1 acetonitrile and H2O and column temperature was set at 40.0°C. The run time was 5 minutes and the solubility was calculated from a standard curve.
Prediction of iJAK-001 lung to plasma partition ratio (Kpu) using SimCYP
Physicochemical and in vitro ADME (absorption, distribution, metabolism and excretion) parameters of iJAK-001 used for lung to plasma partition ratio (Kpu) prediction are summarized in Table 1. Two Kpu values were calculated in SimCYP Rat (version 16.1) based on two published methods, Rodgers and Rowland's method(19) and Poulin and Thiel's method.(20)
Measured Physicochemical Properties of iJAK-001 Used in SimCYP Rat V16 to Predict Lung to Plasma Partition Ratio (Kpu)
MDCK, Madin-Darby canine kidney cells.
Tissue binding of iJAK-001 in rat and human lung homogenate
Samples of lung tissue from rat and human were provided by Bioreclamation IVT (New York, NY). Lung homogenate was prepared as a 4 × dilution in sodium phosphate buffer, pH 7.4, using a SPEX Sample Prep 2000 GENO/Grinder (Metuchen, NJ). Measurement of fraction unbound in the lung homogenate was determined by a rapid equilibrium dialysis device (Thermo Fisher Scientific, Waltham MA) in accordance with the manufacturer's procedures. iJAK-001 was spiked into lung homogenate and dialyzed against buffer for 6 hours. Concentrations of the unbound fraction on the buffer side and drug total from the plasma side were measured by LC-MS/MS (liquid chromatography tandem-mass spectrometry) and calculated from two corresponding standard curves. Extrapolation of fraction unbound in lung (fu, lung) was then calculated from the diluted matrix using the following equation(21):
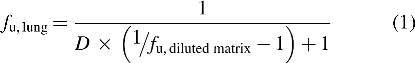
where D is the dilution factor.
Animals
Male Brown Norway rats, weighing 225–250 g (Charles River Laboratories, Kingston, NY), were used for the Alternaria alternata model. For pilot IV/IT PK (pharmacokinetics), experiments were carried out in the jugular vein and carotid artery cannulated Wistar Han rats. All procedures were approved by the Animal Care and Use Committee of Merck & Co., Inc. (Boston, MA) and carried out at Merck & Co., Inc. All experiments adhered to the “Guide for the Care and Use of Laboratory Animals”(22a) (8th edition, NIH publication).
Compound formulation
For IT administration in rats, a mixture of micronized iJAK-001 and the vehicle, phosphate-buffered saline (PBS) containing 0.5% polysorbate 80, was sonicated using a Covaris ultrasonicator (Woburn, MA) to give suspensions. The particle size distribution of the suspended material was evaluated by dynamic light scattering, using a Horiba LA-750 (Horiba Scientific, Piscataway, NJ) with a water background, and the crystal phase of iJAK-001 was confirmed by X-ray powder diffraction after formulation.
For dry powder inhalation delivery in sheep, blends of micronized iJAK-001 and anhydrous cured lactose were prepared. The two components were combined in amber bottles and blended via acoustic mixing on a Horiba LabRAM mixer (Horiba Scientific, Piscataway, NJ). The resulting blend was pushed through a 300 μm sieve and collected to give the final free flowing blend. Content uniformity was measured by UPLC analysis of samples from the top, middle, and bottom of the bottle.
IT iJAK-001 administration
IT instillation was performed based on a modified experimental procedure.(22b) Very briefly, rats were anesthetized with 4% isoflurane supplemented with 100% oxygen. Once anesthetized, the animals were suspended in a supine position on a dosing rack. The larynx was then visualized with a laryngoscope and a 22G 1.25 inch mouse gavage needle, attached to a 1 mL syringe, was inserted into the trachea. Either the vehicle or the iJAK-001 suspension (200 μL or ∼1 mL/kg) was slowly delivered to the lungs. After dosing, animals were returned to their home cage and laid in a supine position until they regained consciousness.
A. alternata model of lung inflammation and IT iJAK-001 administration
The A. alternata model was validated based on a published procedure.(23) Additional details on the model can be found within. Very briefly, male Brown Norway rats (n = 8) received lung challenges of either A. alternata or PBS. Animals were treated with either iJAK-001 or the vehicle before the lung challenge. iJAK-001 or the vehicle was administered via IT delivery either 1 hour or up to 48 hours before challenge and then every 24 hours for the former (QD) or single dose for the latter until study termination, unless otherwise stated. Two days (48 hours) after the challenge, animals were euthanized with sodium pentobarbital and bronchoalveolar lavage fluid (BALf), lungs, and plasma were collected. For collection of BALf, a tracheal catheter was inserted and the lungs were lavaged with two flushes of 3 mL each of 0.9% saline solution. The typical volume returned from these flushes was 4 mL. BALf was then analyzed for inflammatory cell infiltration using Advia 120 Automated Hematology Analyzer (Siemens, Washington, DC). Lung and plasma were collected for pharmacokinetic analysis.
A. alternata extract (Lot# 224126) was purchased from Greer Laboratories (Lenoir, NC). A. alternata was formulated at 10 mg/mL in PBS and 100 μL of the solution was delivered IT via a PennCentury Model IA-1B-R MicroSprayer Aerosolizer (Philadelphia, PA).
Lung and plasma pharmacokinetic profiling
Male Brown Norway rats were dosed IT with iJAK-001 in suspension at 0.03, 0.1, 0.3, and 1.0 mg/kg. Groups of rats (n = 4) were euthanized with sodium pentobarbital at 0.08, 2, 6, or 24 hours following dosing. Longer time points (48 and 72 hours) were also collected after a single 1 mg/kg IT dose. Lungs and plasma were collected for pharmacokinetic analysis.
Sample preparation and LC-MS/MS conditions
Lung samples treated with iJAK-001 were diluted (1:4 weight:volume in water) and homogenized with a GENO/Grinder (Metuchen, NJ) at a rate of 20/min for 5 minutes. An aliquot (50 μL) of plasma or homogenized lung solution was taken for analysis in a 96-well plate format. A single-step protein precipitation was performed by adding 200 μL aliquot of acetonitrile containing an internal standard (IS) mix (a cocktail mix containing diclofenac, imipramine, and labetalol) to standard quantitative controls and biological samples. Plates were mixed and followed by centrifugation at 1950 g for 5 minutes. The supernatant was transferred into a fresh 96 2 mL-deep plate and injected with 5 μL into the LC-MS/MS system. Two separate standard curves, one using blank lung homogenate and the other plasma, were prepared to account for matrix effect during analysis.
LC-MS/MS analysis was carried out on a Waters Acquity UPLC (Waters Corp, Milford, MA) coupled with an AB Sciex QTRAP 5500 mass spectrometer (Framingham, MA) using an ESI (electrospray ionization) source. Reverse-phase chromatography was performed on a Waters Acquity HSS T3 column (2.1 mm × 50 mm, 1.8 μm) at room temperature with gradient elution of solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid). The gradient starts with 5% B for 15 seconds, followed by a linear gradient to 95% B over 1.75 minutes, held at 95% B for 0.5 minutes, and re-equilibrating the column with initial condition of 5% for 0.5 minutes. The flow rate was 0.75 mL/min and the total run time was 3 minutes. The multiple-reaction-monitoring mode was used for analyte detection. Ions were created in positive ion mode with the ion source temperature at 500°C, nebulizer gas with nitrogen (Gas 1) of 50, auxiliary gas (Gas 2) of 60, and curtain gas of 40. The dwell time for each transition was set at 25 milliseconds with an interchannel pause time of 5 milliseconds. The Analyst 1.6 software (Applied Biosystems) was used to control the MS-MS (tandem mass spectrometry) system and for data analyses. Peak area ratios of the target analyte to IS were used for construction of the calibration curve ranging from 0.0001 to 10 μM and quantification of the analyte in biological samples.
Ascaris-sensitive sheep model
A detailed description of the protocols for measuring airway mechanics and aerosol delivery of antigen in the Ascaris-sensitive sheep model can be found in the literature.(24) On the day of antigen challenge, baseline values of pulmonary resistance (RL) were obtained, and then, three sheep were treated with IJAK-001 either via inhalation or intravenous (IV) infusion. For inhaled dosing, 10 mg per animal of iJAK-001 was delivered 1 hour before the aerosolized Ascaris suum. Average weight of the sheep used in the study was about 50 kg. Inhaled iJAK-001 was delivered as a 50:50 blend with anhydrous lactose via a Spinhaler, which was connected to the Harvard pump similarly to the nebulizer described by Moy et al.(24) For the IV infusion dosing, a 0.002 mg/kg bolus injection was given via a jugular catheter 1 hour before challenge, followed by a 0.0012 mg/kg/h infusion for 8 hours. Measurements of RL were obtained 5–10 minutes after injection, to ensure there was no adverse effect of the drug itself on RL. Measurements of RL were obtained immediately after challenge, hourly from 1 to 6 hours after challenge, and on the half-hour from 6 1/2 to 8 hours after challenge. Throughout the study, plasma samples were collected via a jugular vein catheter for analysis of drug concentrations.
Estimation of average local lung concentration
For compound with low solubility, local dissolved average lung concentration was estimated based on a modified equation for intestinal drug concentration calculation commonly used in drug/drug interaction predictions,(25–27) except the blood flow to the intestine was replaced by lung blood flow (Qlung), which is similar to the cardiac output or 74 mL/min for rat, 5600 mL/min for human(28):
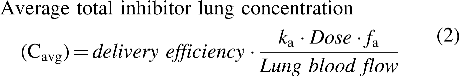
where delivery efficiency is the fraction of the dose reaching the lung after inhalation. ka is the rate of absorption and fa is the fraction absorbed in the lung after inhalation. Rate of absorption was estimated based on the mean resident time (MRT) principles(29) or published literature(30):
fa was calculated based on the dose-normalized systemic AUC (area under the curve) ratio between IT and IV delivery. For discussion purpose throughout the article, the density of lung homogenate was assumed to be 1 or 1 g of tissue = 1 mL.
Statistical analysis
All inflammation endpoints were analyzed with a one-way analysis of variance, with post hoc comparisons being done using Bonferroni's multiple comparison test. A p-value of <0.05 was considered to be statistically significant. All results are expressed as mean ± standard error of the mean (SEM). The SEM is based on the observed variability in the treatment groups.
Results
Kinetic and thermodynamic solubility of iJAK-001 dry powder in SLF
Because of the technical challenge of measuring directly the lung surface concentration of solid particles deposited, an experimental model was designed to fix the amount of iJAK-001 at 1 mg and suspend in progressively lower amount of SLF (Table 1) to extrapolate in theory the limited fluid environment on the surface of the lung. Solid particles with 1–5 μm diameter of iJAK-001 showed limited dissolution in SLF and at 5 minutes the solubility already reached a maximum of 2–4 μg/mL (∼5–10 μM, molecular weight of iJAK-001 = 411 μg/μmol). iJAK-001 did not show increased or diminished solubility over 24 hours, suggesting dissolution was complete. Decreasing the amount of SLF did not affect the solubility up to 3 mg/mL (nominal concentration) of iJAK-001, indicating that the fraction dissolved decreased with an increasing amount of the inhibitor.
Prediction of lung to plasma partition ratio of iJAK-001 using SimCYP
The measured physicochemical properties of iJAK-001 are summarized in Table 1. Because the vegetable oil/buffer partition ratio at pH 7.4 (Log Dvo:buffer) was not available experimentally, the predicted value of 0.78 from octanol/water partition ratio was used in the simulation (i.e., Log Po:w was assumed to be the same as Log Dvo:buffer). Based on these parameters, the predicted lung to plasma partition ratio (Kpu) is 0.60 using the method of Rodgers and Rowland or 0.8 using Poulin and Thiel's method.(19,20)
Measurement of fraction unbound (fu, lung) of iJAK-001 in rat and human lung homogenate above or below the thermodynamic solubility limit
The measured unbound plasma and lung homogenates in rat and human are summarized in Table 2 and ranged from 0.089 to 0.16. No concentration-dependent change in fu, lung was observed. Very little difference in binding between rat and human was observed. Plasma protein binding of fluticasone propionate in human was used as control and the fu, plasma obtained (0.012) is consistent with that reported in literature, 0.016.(31)
Unbound Fraction of iJAK-001 Above or Below the Thermodynamic Solubility of 5 to 10 μM in Rat and Human Lung Homogenate After Rapid Equilibrium Dialysis
Measurement of concentration of iJAK-001 and tofacitinib in the lung and plasma of rats over time
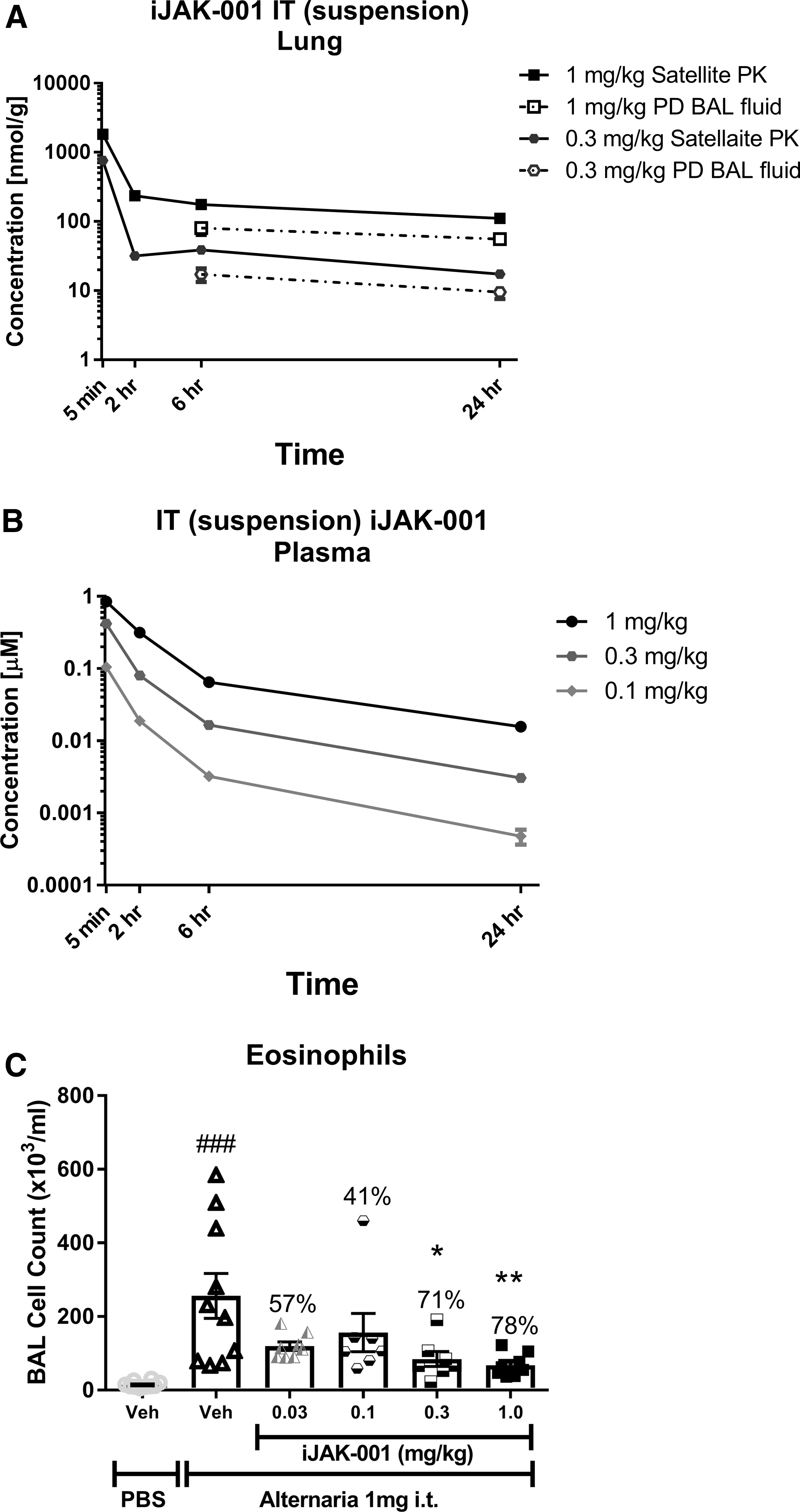
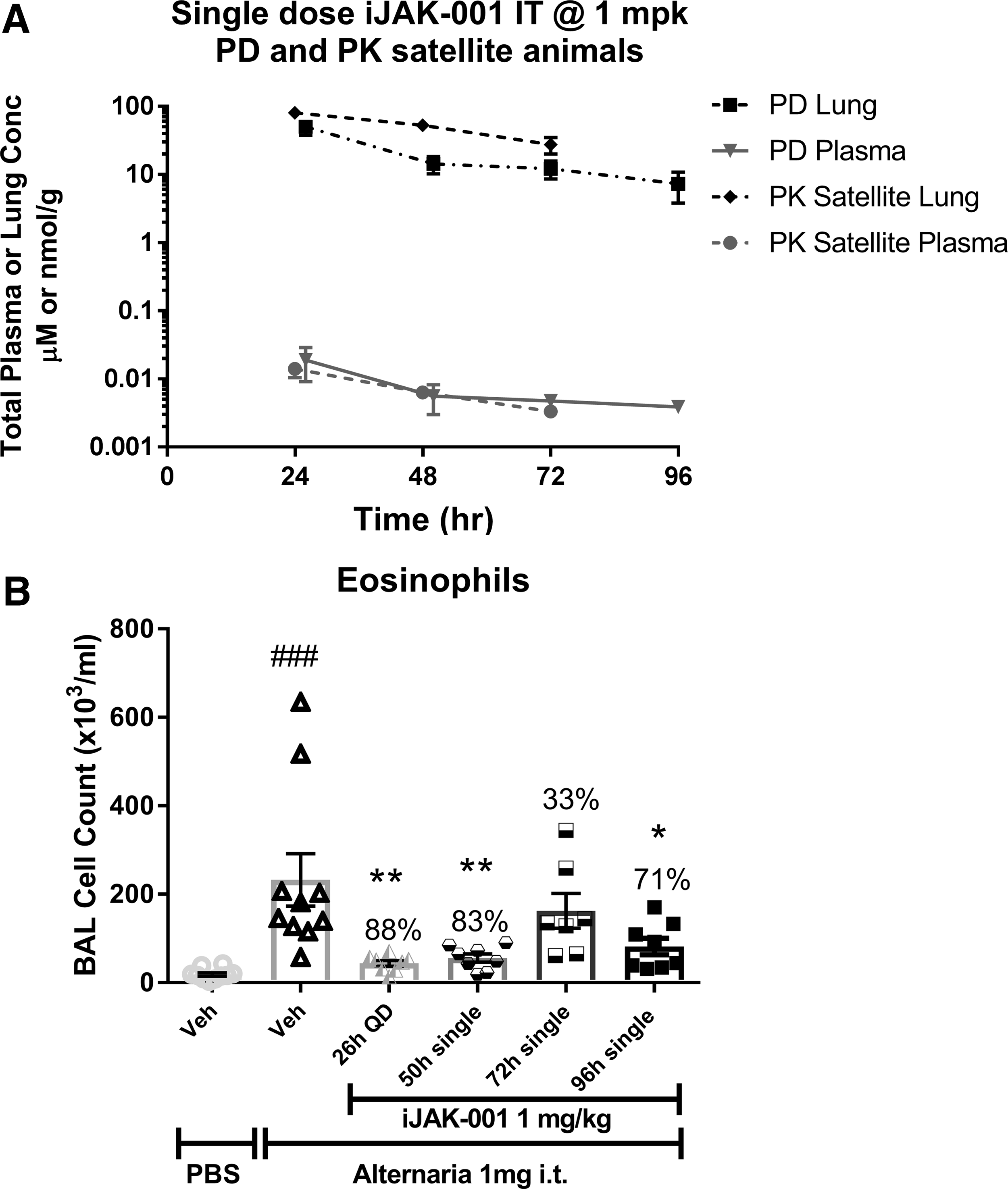
iJAK-001 in suspension was dosed IT at multiple single doses (0.03, 0.1, 0.3, and 1.0 mg/kg) in male Brown Norway rats. After dosing, lungs (Fig. 1A) and plasma (Fig. 1B) from the 0.3 and 1.0 mg/kg groups were collected at 5 minutes, 2, 6, and 24 hours postdose and compound concentrations were measured in those tissues. As shown in Figure 1A, the lung concentration (Cmax) of iJAK-001 peaked immediately after 0.3 and 1 mg/kg doses (761.8 ± 82.7 and 1822.4 ± 100.9 nmol/g, respectively). A relatively flat distribution phase followed with measureable concentrations still found 24 hours postdose in the lung, indicating the compound levels equilibrated 2 hours after dosing. In plasma (Fig. 1B), the PK profile of iJAK-001 after IT dosing had two distinct phases, the first was a rapid absorption phase followed by a slow “depot” release phase that lasted for hours. Due to recovery from anesthesia after IT dosing, Cmax was at the first time point, 5 minutes. It should be noted that assuming density of lung homogenate = 1, after 5 minutes the lung concentrations were 1816- and 2146-fold higher than measured in plasma for the 0.3 and 1.0 mg/kg doses, respectively. A linear dose-proportional increase in AUClung, 0 to 24 hours (Table 3) was observed for the two dose groups. In contrast, a dose-dependent systemic (plasma) bioavailability was observed that ranged from 100% at 0.1 mg/kg to 28% at 1 mg/kg indicating that the fraction absorbed decreased with increasing dose (Table 3). However, the 28% bioavailability at 1 mg/kg is likely an underestimation because iJAK-001 was not completely cleared from the lungs over the course of 24 hours. To improve the overall estimation of lung or plasma AUC in a separate experiment, a single IT 1 mg/kg dose was evaluated over the course of 96 hours to get a better idea of the retention of iJAK-001 in the lung. iJAK-001 was present in the lung even after 96 hours of dosing (7.4 ± 3.5 nmol/g; Fig. 2A) and the partitioning between lung and plasma was maintained with lung concentrations being several thousands of fold higher than in the plasma. Indeed, lung AUC0–∞ based on 0–24 hours of lung exposure increased by 11% from 0.55 to 0.61 μM × h after calculating AUC0–∞ based on 0–96 hours of exposure. In addition, based on the terminal lung concentrations from PK animals up to 72 hours postdose, the disappearance of iJAK-001 was linear and followed a zero-order disappearance with linear regression coefficient (r2) of 0.999, slope = −1.1, and intercept = 106.8 μM.
Plasma and Lung Exposure of iJAK-001 at Various Doses After IV Bolus and Intratracheal (Suspension) Dosing in Wistar Han Rat
Total dissolved and undissolved drug. Only lung lobes were collected and weighed. Trachea collection was avoided for ease of tissue homogenization.
AUC, area under the curve;
For comparison, the PK of a 1 mg/kg dose of tofacitinib were evaluated, a highly water soluble JAK inhibitor, after IT dosing in both matrices (Fig. 3A). At this dose, tofacitinib had a peak concentration of 8.4 ± 0.4 nmol/g in the lung measured 5 minutes after dosing and was cleared completely from the lung within 4 hours. At an equivalent dose, tofacitinib showed a much lower lung to plasma partitioning than iJAK-001, with lung concentrations being only 4.9-fold higher than plasma at the first time point and over time, this partition ratio approached unity. Taken together, these data indicate that the physicochemical properties of iJAK-001 lead to improved lung retention and a greater lung to plasma ratio than the more water soluble JAK inhibitor, tofacitinib.

Evaluation of iJAK-001 and tofacitinib in the Alternaria-induced model of rat lung inflammation
The A. alternata model of lung function was used to evaluate the PD effects of iJAK-001 after IT delivery. The ability of iJAK-001 to inhibit eosinophil infiltration after A. alternata challenge at multiple doses (0.03, 0.1, 0.3, and 1.0 mg/kg, QD) was evaluated (Fig. 1C). iJAK-001 inhibited eosinophil recruitment dose dependently (57% ± 5%, 41% ± 21%, 71% ± 8%, and 78% ± 5%, respectively), with the 0.3 and 1.0 mg/kg doses being significantly different from the vehicle-treated and A. alternata-challenged control group. For comparison, tofacitinib was evaluated in the A. alternata model (Fig. 2B). However, due to its rapid clearance from the lung, the compound was dosed BID at daily doses of 1.6, 5.0, and 15.0 mg/kg/dose. In that study, a 1.6 mg/kg dose of tofacitinib had no effect on eosinophil infiltration (14% ± 14% inhibition), while a 15.0 mg/kg BID dose completely inhibited eosinophil infiltration (105% ± 3% inhibition; p < 0.001, versus vehicle without Alternaria challenge). The 5.0 mg/kg BID dose gave equivalent inhibition to the 1.0 mg/kg QD dose of IJAK-001 (78% ± 8% vs. 82% ± 5%, respectively; p < 0.001, vs. vehicle without Alternaria challenge for both). Finally, the duration of action of iJAK-001 was evaluated by varying the pretreatment time before allergen challenge and holding the dose constant at 1.0 mg/kg (Fig. 3B). For three groups, a single dose of iJAK-001 was delivered at 2, 24, or 48 hours before the challenge with A. alternate (50, 72, and 96 hours postsingle IT dose). For the control group, the same QD dosing schedule was maintained as in previous studies which again significantly inhibited eosinophil infiltration (88% ± 3%; p < 0.01, vs. vehicle without Alternaria challenge). A single dose delivered 2 hours (83% ± 4%; p < 0.01, vs. vehicle without Alternaria challenge) or 48 hours (71% ± 9%; p < 0.05, vs. vehicle without Alternaria challenge) before the challenge also inhibited the inflammatory response. Interestingly, the dose delivered 24 hours before the challenge led to weaker inhibition (33% ± 18%; not significant) than either of the other single-dose groups. Nevertheless, these data indicate that iJAK-001 is a potent molecule with activity that lasted at least 48 hours after dosing if not longer in our in vivo rodent model.
Lung to plasma partitioning of iJAK-001 after solution IV, IT, and IT suspension
After IT dosing of iJAK-001 in suspension, the total concentration of iJAK-001 in the lung (density assumed = 1) was around a 1000-fold greater than in the plasma, depending on the dose delivered. These data suggested that there was significant partitioning between the two compartments, but it is well known that lung concentration measurements could be erroneously skewed by the undissolved portion of compound.(13,14) To understand the true partitioning between lung and plasma, iJAK-001 was delivered in different formulations. As shown in Figure 4A, after a 0.25 mg/kg IV dose of iJAK-001, there was no difference in the lung and plasma concentrations. A new formulation of iJAK-001 was tested that put the compound in a solution and was delivered a 0.3 mg/kg dose of iJAK-001 IT (Fig. 4B). The lung concentration 15 minutes after dosing was 38-fold higher than in the plasma, but this difference diminished over time and by 4 hours after dosing the concentrations in the two tissues had equilibrated. In addition, very little compound was detected after 4 hours, suggesting fairly rapid elimination after solution delivery.
Topical application of iJAK-001 inhibits lung function changes in the Ascaris-sensitive sheep model
The A. suum-sensitive sheep model was used to evaluate the effects of iJAK-001 on lung function when delivered as an inhaled dry powder as opposed to the suspension delivered in the rat model. A 10 mg dose of iJAK-001 (Fig. 5A), delivered in a 50:50 blend with lactose 1 hour before A. suum challenge, inhibited the increase in airway resistance both during the Early Airway Response (45% ± 4%; p < 0.001, vs. vehicle 30 minutes after challenge) and the Late Airway Response (90% ± 3%; p < 0.01, vs. vehicle 7 hours after challenge). After inhaled dosing, iJAK-001 was detectable in the plasma of sheep for the next 6 hours with an AUC0–8 hours of 0.007 μM h. To confirm that the activity seen with iJAK-001 was due to the topical administration, iJAK-001was dosed via IV infusion (Fig. 5B). iJAK-001 was delivered with an IV bolus loading dose of 2 μg/kg/h before the A. suum challenge, followed by a slow infusion of 1.2 μg/kg/h for the next 8 hours to match the systemic exposure seen in the first study. When delivered this way, iJAK-001 had no effect on either the Early or Late Airway Response even though average plasma concentrations were in the same range.
Comparison of predicted human average lung concentration and observed clinical lung exposure of ICS
To validate the usefulness of Equation (2), the average total inhibitor lung concentration (Cavg) was calculated and summarized in Table 4 for fluticasone propionate, ciclesonide, and budesonide based on measured clinical lung exposures,(15,32,33) physicochemical properties, and PK parameters,(12) including aqueous solubility, inhaled dose, human plasma clearance after IV, systemic bioavailability after inhaled or oral dose, and rate of absorption. For rate of absorption, fluticasone propionate was published(30) and this value was assumed to be the same for ciclesonide and budesonide. Fraction of the dose reaching the lung was estimated based on the difference between inhaled and oral bioavailability (%Finhaled − %Foral). Because of the heterogenicity of lung distribution after inhalation of corticosteroids in dry powder, all the reference articles published the mean, the median, or the range of drug exposure in both central and peripheral lung tissues. They were then compared with the estimated Cavg. Overall, high interindividual variability was observed for all three inhaled drugs. Fluticasone propionate at 1 mg inhaled dose, for example, in one study, the exposure ranged from below quantitation limit to 45 pmol/g of tissue over the course of 22 hours in the central lung to a spread of BQL to 7 pmol/g in the peripheral lung. In another study, this value ranged from a mean of 87 pmol/g (BQL to 459 pmol/g) at Cmax in the central lung to a mean of 18 pmol/g (2–40 pmol/g) in the periphery Cmax. For both ciclesonide and budesonide, lung exposure of unchanged parent drugs and their metabolites was published, but for simplicity, only the parent drug concentrations were summarized. Because of the high interindividual and interstudy variabilities, no attempts were made to estimate measured Cavg from the PK data.
Comparison of Predicted Local Average Lung Concentration (Cavg) Using Equation (2) as Described in Materials and Methods and Observed Central and Peripheral Human Lung Tissue Concentrations After Inhalation of Fluticasone Propionate, Ciclesonide, and Budesonide
Cavg estimated from ka·Dose·(%Finhaled − %Foral)/Lung blood flow; density of lung tissues is assumed to be 1 (1 g = 1 mL).
BQL, below quantitation limit.
Discussion
Estimation of local lung concentration after inhaled delivery of a poorly soluble drug has always been a challenge. After inhalation of solid particles, to ensure good recovery of drug from lung homogenate, standard bioanalytical procedures typically call for a strong organic solvent extraction. However, both the homogenization of lung tissue and the solvent extraction may solubilize undissolved dose deposited in the lung. Thus, one could erroneously use the total lung burden (dissolved plus undissolved fractions) to establish PKPD relationship in drug discovery.(34) Similar to the free-drug hypothesis, it is well accepted that only the dissolved portion of the dose deposited in the lung is available for absorption. In addition, the heterogeneously distributed solid particles in the lung are homogenized and averaged during tissue preparation, and therefore, the concentration obtained is not a good reflection of the local concentration of drug along the lung epithelial lining. Furthermore, the total fluid volume of lung is about 0.045 to 0.055 mL in rat and 10–40 mL in human. The average lung tissue weight is about 1.5 g in rat and 1000 g in human.(35) Therefore, normalization of drug amount by tissue fluid volume, tissue weight, or tissue surface area would give very different lung concentration units. It is unknown at this point which concentration unit would provide the best estimate of efficacious lung concentration needed to establish a PKPD relationship. Alternatively, it is possible to collect BALf after inhalation. However, due to the heterogeneity of dose delivered to the lung, caution has been advised against this approach.(13,14) For example, introduction of wash fluid during bronchoalveolar lavage (BAL) collection might either solubilize or wash away some of the deposited lung dose along the upper airway (Fig. 1B). Twofold lower lung concentrations of iJAK-001 were consistently seen after BALf collection, but the corresponding systemic plasma concentration was not affected, indicating that the change in lung concentration is mainly from undissolved fraction. In addition, circulating plasma concentration of test compound may not be a good representation of local dissolved concentrations at the lung epithelial lining after inhalation (as outlined below). This is also true for drugs that have unbound lung to plasma partition ratio (Kpuu) significantly greater than one because of uptake transporter(s) or for basic drugs that are known to be trapped in the cellular lysosomal compartment.(36) To minimize these uncertainties, multiple indirect methods are needed to estimate the fraction dissolved and the average local lung concentration of iJAK-001 after IT delivery. These methods included kinetic and thermodynamic solubility measurement of iJAK-001 at progressively lower amount of SLF (1 mg of solid particles with average particle size ≤5 μm dissolved 0.3 to 30 mL of fluid, equivalent to nominal concentration of 0.3 to 3 mg/mL in Table 1); lung to plasma partition ratio estimation (in silico, in vitro, and in vivo); preclinical PKPD relationship of a highly soluble benchmark pan-JAK inhibitor, tofacitinib; duration of PD response and plasma/lung exposure relationship after single IT dose of iJAK-001; and differences in PD response after steady-state IV infusion of iJAK-001 versus inhalation matching the systemic (plasma) exposures for both in the sheep asthma model.
Based on biopharmaceutical drug classification,(37) iJAK-001 has high permeability and low solubility similar to some of the ICS such as fluticasone propionate (Table 1). The results from in vivo studies demonstrated that neither average total lung concentration measurement nor systemic plasma concentration of iJAK-001, after IT delivery, was a good reflection of the local lung epithelial lining concentration because of two key reasons. The first reason, after IV solution dosing, the free lung to plasma ratio (Kpuu) is approximately one confirming that there is no preferential uptake of iJAK-001 into the lung tissue (Fig. 4A, steady state assumed). In contrast, when iJAK-001 was dosed in suspension, the lung to plasma partition ratio was 1000-fold (Fig. 4C, equilibrium assumed), thus supporting that the AUC difference between the two dosing routes is the fraction undissolved. These data also indicated that the amount of compound that was dissolved and thus available for pharmacological activity was likely far lower than the measured values in our earlier in vivo PD studies (i.e., IT using suspension formulation). Because of the uncertainty in the dissolved lung concentration of iJAK-001 after IT delivery, the inhibitor lung concentrations at different doses were estimated using Equation (2), which has been used successfully for intestinal drug/drug interaction predictions,(25–27) and are summarized in Table 5. For IT delivery, the fraction reaching the lung was assumed to be one or delivery efficiency of 100%. The rate of absorption ka was estimated to be one after correcting for the MRT difference between IT and IV delivery (Supplementary Data). This rapid rate of absorption is also consistent with the short Tmax (first time point or 5 minutes) after IT dosing. Finally, because dose-dependent bioavailability was observed, from 100% at 0.1 mg/kg to 28% (for ease of comparison, this was extrapolated from AUC0–∞ based on 0–24 hours of exposure of all doses) at 1 mg/kg, fa was estimated to be similar to bioavailability because systemic bioavailability approached unity at the lowest dose, indicating that absorption was complete with little or no lung metabolism taking place. Based on these numbers, the average inhibitor concentrations (total and unbound) along the lung epithelial lining at different doses were calculated and are summarized in the same table.
Estimated Epithelial Lung Surface Concentration in Rat After Intratracheal Delivery of iJAK-001 in Suspension Using Equation (2)
iJAK-001 was stable to lung S9 metabolism, so fa is similar to bioavailability. In addition at lowest dose (0.1 mg/kg), systemic bioavailability was one suggestion, fa is also approaching unity. A dose-dependent absorption (fa) was observed. ka was estimated based on mean resident time principles [Equation (3) in Materials and Methods] and it was close to 1.
The second reason is that using the systemic plasma concentration after inhalation, such as Cavg, for PKPD or human dose projection, will likely result in erroneous estimation of the in vivo potency. This is evident based on two key experiments: the reduction in sheep early and late airway resistance after allergen challenge was only evident after inhalation of iJAK-001 dry powder at 10 mg per animal. In contrast, when iJAK-001 was infused IV to steady state in sheep, and the plasma exposure was matched to that following inhalation, there was no change in PD response. In the second experiment, after a single dose of iJAK-001 at 1 mg/kg IT in rat, up to the maximal PD response was observed, which lasted for a few days after delivery despite total Cavg plasma concentrations of single nM concentration, which is significantly lower than iJAK-001's cellular potency achieved at 50, 72, and 96 hours postsingle dose (Table 6). This contrasts with a lack of PD response in rat at 0.1 mg/kg IT QD dose (total Cavg in plasma was also around single digit nM at this dose, Table 3). These data indicate that the activity seen in the preclinical models with iJAK-001 was dependent on direct delivery to the lungs rather than any leakage into the periphery. Therefore, local average inhibitor concentration, together with fraction unbound in lung, is more appropriate for human dose projection. This is also in agreement with the pathophysiology of an asthma attack. That is, allergen- or pollutant-induced airway inflammatory infiltrates at the contact surface such as eosinophils, lymphocytes, and mast cells can cause recruitment of cytokines leading to abnormal immune (Th2) response localized in the lung rather than systemic response.(38) However, loss of statistically significant PD response 72 hours postsingle dose in rat versus other time points (33% inhibition; Fig. 3B) might be due to either poor delivery technique or heterogeneous distribution of the compound in the lung. They should be considered when frequency of dosing prediction in human is needed as variability in dose/response might increase over time after single dose.
Lung Burden (Dissolved + Undissolved Fraction) and Plasma Total and Unbound Exposure of iJAK-001 After 1 mg/kg Single Dose Intratracheally (Suspension) in Brown Norway Rats
Total dissolved + undissolved fractions.
PK, pharmacokinetics.
Additional caution must also be exercised using Equation (2) as the oral exposure (AUC) from the fraction swallowed (foral) after inhalation of dried powder must be subtracted before making such estimation. Therefore a correction factor or delivery efficiency was added to Equation (2). Traditionally, estimation of the fraction that reached lung is typically obtained from lung deposition imaging studies using radiolabeled technetium-99 m(39) or oral versus inhaled PK studies with charcoal codosing experiments.(40) For iJAK-001 preclinical studies, no correction from oral absorption (fa, oral) was included because delivery efficiency was assumed to be 100% after IT. For dried powdered delivery of ICS in human, fraction absorbed in the lung [delivery efficiency × fa from Equation (2)] was estimated from the difference between percent pulmonary bioavailability (%Fpulmonary) and oral bioavailability (%Foral) reported in literature.(12) Based on published values for ICS, delivery efficiency can range from 10% to 80%.(12) Therefore, more in-depth clinical study to estimate the epithelial average concentration of iJAK-001 by knowing the %Fpulmonary and %Foral in human will be needed. Alternatively, they may be estimated from single-species allometry.(29) Despite species differences in the total lung fluid volume (0.045–0.055 mL in rat to 10–40 mL in human), the solubility of iJAK-001 at 1 mg in different volumes of SLF (from 0.3 to 30 mL or from 0.3 to 3 mg/mL nominal concentration in Table 1) did not change over 24 hours, suggesting that there will be little or no species difference in the fraction dissolved after dose normalization between rat and human. However, in rat, a dose-dependent bioavailability of iJAK-001 was observed after IT dosing at 0.1 to 1 mg/kg, suggesting that the fraction absorbed decreased with increasing dose. More research will be needed to estimate fraction absorbed in human after inhalation of this potent inhibitor.
To further demonstrate the importance of solubility and lung retention, tofacitinib was also dosed IT (solution) in vivo. Because of its high aqueous solubility (>150 μM in phosphate saline buffer, pH 7.4, in-house data), it was not possible to suspend the drug in a water-based formulation. After IT dosing in a solution formulation, it was evident that there was no depot effect in the lung (Fig. 2A). The drug distributed rapidly throughout the systemic circulation and cleared (total CLrat, in vivo = 39–76 mL/min/kg). From preclinical and clinical PKPD studies conducted in a rheumatoid arthritis model in mouse and in patients,(4) it was suggested that 8–12 hours of JAK IC50 Cavg, free coverage was needed for efficacy. Therefore, tofacitinib was dosed BID in our asthma model. Furthermore, free tissue to plasma partition ratio for tofacitinib was also estimated to be close to one. Therefore, plasma concentration is a good reflection of the average lung concentration for the drug because fraction dissolved is 1. A similar observation can be made about iJAK-001 when it was given IT in a solution formulation after equilibration (i.e., fairly rapid elimination in lung with CLrat = 20 mL/min/kg). In addition to poor lung retention, tofacitinib is about fivefold less potent than iJAK-001 in human whole blood JAK inhibition assay (data not shown), but its free fraction in plasma and lung is about threefold higher than iJAK-001. Consequently, tofacitinib had to be dosed at 5 mg/kg BID IT to have a comparable PD response as 1 mpk single dose IT of iJAK-001 (78% vs. 82%, respectively).
In conclusion, establishing a proper PKPD relationship of poorly soluble molecules delivered by inhalation continues to be a scientific challenge. To further demonstrate this, predicted Cavg of fluticasone propionate, ciclesonide, and budesonide in human from different inhalation studies based on Equation (2) is summarized in Table 6. Experimentally, because of the heterogeneity of lung distribution (e.g., central vs. peripheral lung concentration differences), low solubility of dried powder (e.g., fluticasone propionate and ciclesonide), and high interindividual variability (e.g., more than two orders of magnitude in lung exposure observed for ciclesonide and its metabolites), it is difficult to validate this equation until a better bioanalytical method can be developed to separate undissolved from dissolved fraction because the equation assumes homogeneous distribution and complete dissolution of solid particles in the lung after inhalation. Ciclesonide, for example, has very high clearance (54 mL/min/kg) in human and yet when it was delivered inhaled as dried powder, lung tissues showed significant depot effect that lasted for almost 24 hours after dosing, suggesting that the lung concentration of the parent drug was not completely dissolved. Therefore, development of a target engagement tool in the clinic such as PET (positron emission tomography) tracers will be needed for accurate prediction of lung delivery and fraction absorbed. In the absence of such a tool, the studies described here showed that it is possible to estimate the average total lung epithelial lining concentration by factoring in the delivered IT dose fraction undissolved, absorbed, and rate of absorption, still useful for clinical candidate selection in the discovery stage because advanced PBPK modeling is not possible due to lack of experimentally validated data. This is also supported by multiple indirect preclinical PKPD experiments. iJAK-001 continues to be a strong early development candidate for the treatment of severe asthma.
Footnotes
Acknowledgments
The authors thank Dr. Iain Martin for the helpful discussion, Drs. Donald Tweedie, James Baker, Ghassan Fayad, Guido Hugo Jajamovich, and Balaji Bharatwaj for the insightful editorial comments, Dr. Vincenzo Pucci for the PK analysis, and Ms. Laura Surdi for measuring the free fractions of iJAK-001 in various biological matrices.
Author Disclosure Statement
The authors declare there are no competing financial interests.
Reviewed by:
Jane Kenny
Jim Zheng
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.