
Abstract
Sinonasal anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma (ALCL) without nodal involvement is extremely rare and the rarity of this tumor often leads to diagnostic dilemma. It has been predominantly reported in pediatric, adolescent and young adult patients, mostly of Asian origin. A 21-year-old female patient presented with history of epistaxis for 1 year. On clinical and radiological examination, there was a 5 cm mass in the right nasal cavity, ethmoid, and frontal sinus. Biopsy at a local center had shown moderately differentiated squamous cell carcinoma. Rebiopsy at our center showed possibility of a hematolymphoid malignancy(pancytokeratin-, CD45+, CD3−, CD20−) and further immunohistochemistry studies(CD4+, CD43+, CD30+, ALK+) revealed ALK-positive ALCL. Rest of the lymphoma work-up was essentially normal and she had stage IE disease. She was treated with a combination of four cycles of cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisolone (CHOP) regimen followed by local radiotherapy (36 Gray/20 fractions/4 weeks) by three-dimensional conformal technique. She tolerated the treatment well without any severe toxicity and had complete clinical and radiological response. At last follow-up visit, 40 months from the initial diagnosis, she was alive and disease free. Sinonasal ALK-positive ALCL is a rare tumor, which can be effectively treated with a combination of multiagent CHOP/CHOP-like regimen and local conformal radiotherapy.
Introduction
Sinonasal anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma (ALCL) is an extremely rare tumor entity. Only a few cases of sinonasal ALCL without nodal involvement have been reported in medical literature until date.1,2 This tumor has been reported in pediatric, adolescent, and young adult population, mostly of Asian origin.1,2 Owing to the lack of awareness of this tumor and the numerous other diagnostic possibilities of a sinonasal mass, there is a potential for diagnostic dilemma and delay. 1 However, careful examination of the histopathology specimen and relevant immunohistochemistry tests (particularly CD30 and ALK) help in ascertaining the diagnosis.
The multimodality management of sinonasal ALCL in adults essentially consists of 3–6 cycles of chemotherapy with cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisolone (CHOP)/CHOP-like regimen depending upon the disease stage followed by local conformal radiotherapy with 30–40 Gray in conventional fractionation. 3 We herein describe a case of sinonasal ALK-positive ALCL in a 21-year-old female patient, treated successfully with a combination of four cycles of CHOP regimen followed by local conformal radiotherapy (36 Gray in 20 fractions over 4 weeks).
Case Report
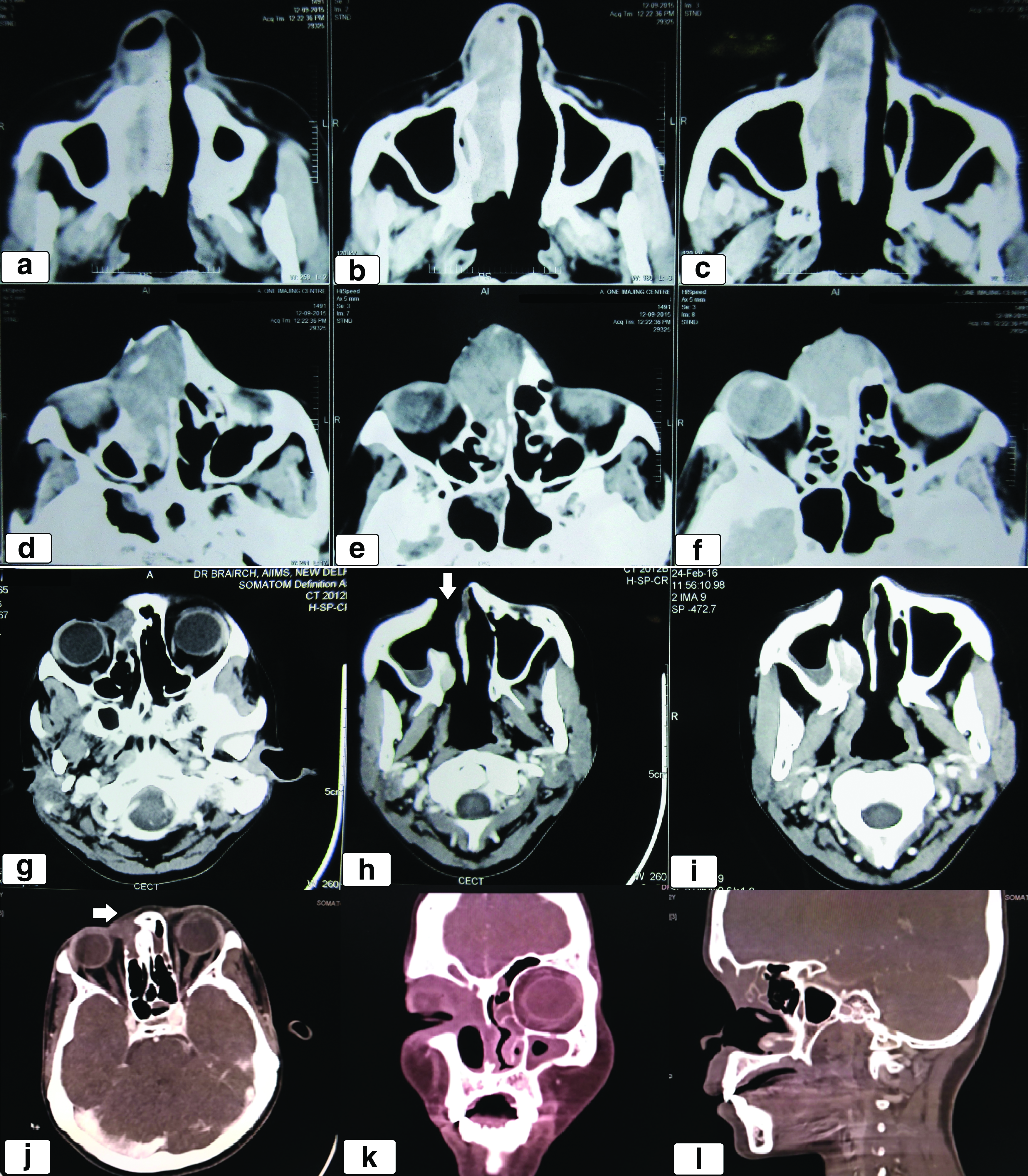
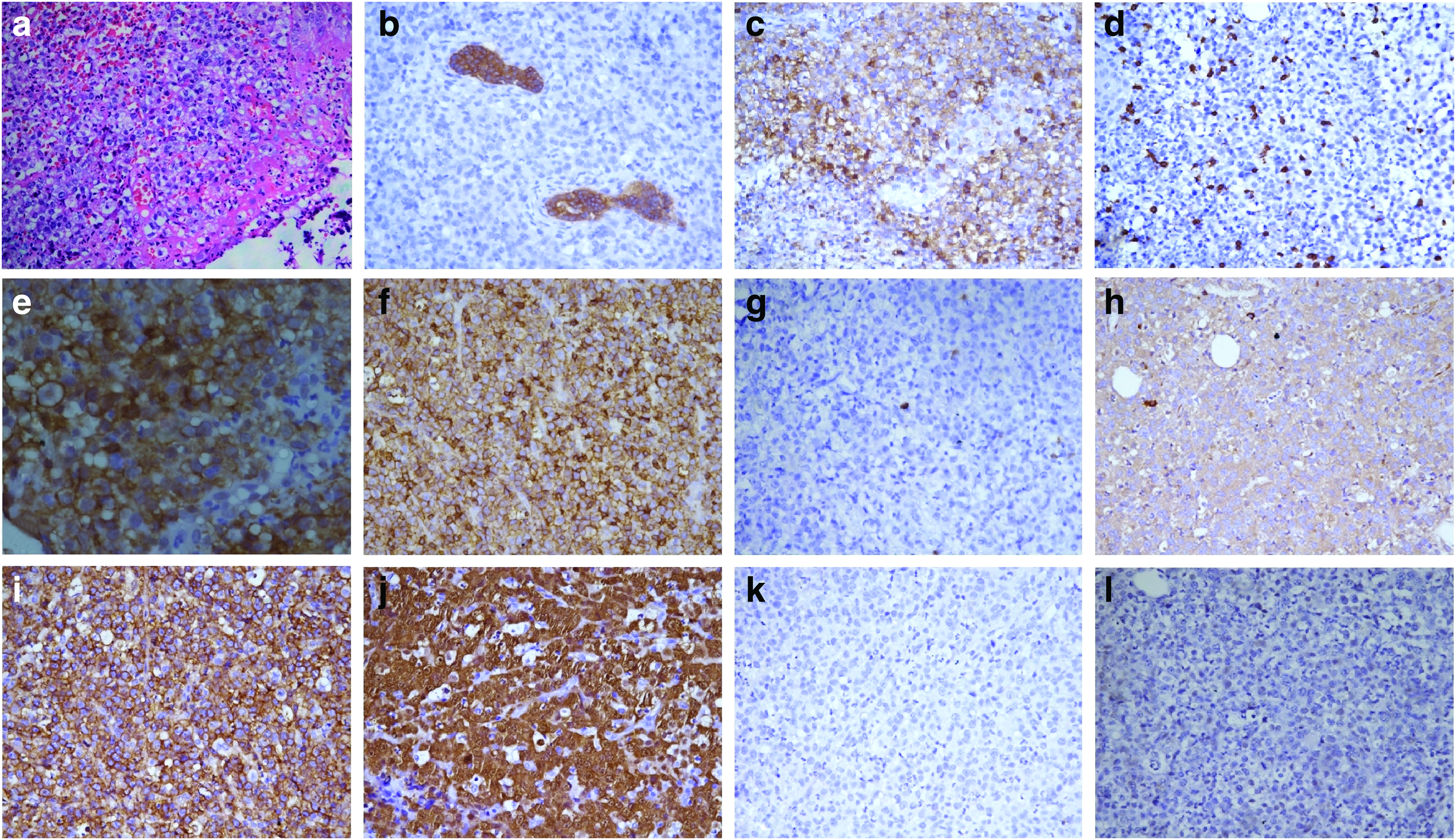
A 21-year-old female patient presented to our clinic with gradual onset, progressively increasing epistaxis for the last 1 year and right nasal stuffiness and difficulty in breathing for the last 6 months. On local examination, there was a 4 × 4 cm ulceroproliferative lesion involving the dorsum of nose and extending to the medial canthus of the right orbit and growth protruding through the right nasal cavity. There was no evidence of any other site of cutaneous involvement. Baseline contrast-enhanced computed tomography (CT) scan of the paranasal sinus showed a 5 × 4 × 2.5 cm minimally enhancing soft tissue density mass involving the right nasal cavity, ethmoid and frontal sinus, eroding the medial wall of the right orbit and the right maxillary sinus with the likely diagnosis of an inverted papilloma (Fig. 1a–f). Nasal biopsy at a local center had shown features of moderately differentiated squamous cell carcinoma (MDSCC). Biopsy from the growth on the dorsum of the nose at our institute revealed ulcerated epidermis with medium-sized cells with clear to eosinophilic cytoplasm and inconspicuous nucleoli along with inflammatory cells and scant necrosis (Fig. 2a). On immunohistochemistry, the tumor cells were immunopositive for leukocyte common antigen, CD43, focally for CD68 and CD117 and immunonegative for pancytokeratin, CD3, CD20, CD79a, PAX 5, Epstein–Barr Virus latent membrane protein, CD138, and myeloperoxidase (Fig. 2). Possibility of a hematolymphoid malignancy was considered. Cellular bone marrow aspirate and biopsy showed normal hematopoietic cells of all series (myeloid:erythroid ratio = 5:1) with no apparent increase in blasts. Serum lactate dehydrogenase was noted to be 226 units/L (normal range 125–220 units/L). On further immunohistochemistry, tumor cells were immunopositive for CD4, CD30, ALK, and CD56 (Fig. 2e, i, and j). Overall features were consistent with ALK-positive ALCL. Chromosomal analysis showed 46, XX with no evidence of structural or numerical abnormality in any of the metaphase. Contrast-enhanced CT scan of the neck, chest, abdomen, and pelvis was essentially normal. The final diagnosis was sinonasal ALK-positive ALCL stage IE. The patient received four cycles of chemotherapy with CHOP regimen (injection cyclophosphamide 750 mg/m2 IV D1, injection adriamycin 50 mg/m2 IV D1, injection vincristine 1.4 mg/m2 IV D1, and tablet prednisolone 100 mg OD D1–D5) every 3 weeks. She tolerated chemotherapy well without any instance of severe toxicity and had significant decrease in the size of the mass (more than 50%). Postchemotherapy CT scan of the paranasal sinus, neck, chest, abdomen, and pelvis showed a heterogeneous soft tissue mass along the lateral wall of the right nasal cavity with extension to the medial canthus of the right orbit. Compared with the baseline CT scan, there was more than 50% decrease in the size of the mass suggestive of partial response (PR) (Fig. 1g–i). Subsequently, she underwent involved field radiotherapy (IFRT) 36 Gray in 20 fractions over 4 weeks. The gross tumor volume (GTV) included the prechemotherapy disease in the right nasal cavity, ethmoid and frontal sinus, and the medial wall of the right maxilla and orbit. The clinical target volume (CTV) encompassed bilateral nasal cavity, ethmoid, frontal sinus, right maxilla, palate, and 5 mm expansion of the GTV in the right orbit. The planning target volume consisted of the CTV with an isotropic expansion of 5 mm (Fig. 3a–c). Radiotherapy was planned by three-dimensional conformal technique (3D-CRT) using anterior, left, and right lateral fields with 6 MV X-rays (Fig. 3d–f). A customized wax bolus was used to provide adequate dose distribution in the superficial region and fill the surface defect in the dorsum of the nose (Fig. 3d). She tolerated IFRT well with minimal adverse effects (grade 1 dermatitis, mucositis, and conjunctivitis). Three months after the completion of radiotherapy, she was clinically disease free with no obvious growth on zero degree endoscopy. Contrast-enhanced CT scan of the paranasal sinus and neck showed minimal soft tissue thickening in the lateral wall of the right nasal cavity extending to the medial canthus of the right orbit suggestive of post-treatment changes (Fig. 1j–l). CT-guided fine needle aspiration cytology from the soft tissue thickening did not reveal any evidence of malignancy. Subsequently, she was on regular follow-up every 3 months. At the last follow-up visit, 40 months from the initial diagnosis, there was no evidence of malignancy on clinical and radiological examination. The acute adverse effects of IFRT had all resolved without the emergence of any late effect of chemotherapy and IFRT. However, there was a defect in the right side of the dorsum of the nose due to the response of the tumor to sequential chemoradiotherapy and she is due for closure of the defect by rhinoplasty.

Discussion
Lymphoma accounts for 5% of all head and neck malignancies. 1 Head and neck lymphomas may be Non-Hodgkin lymphoma (NHL) or Hodgkin lymphoma (HL) with nodal or extranodal involvement. The most common subsite of extranodal lymphoma in the head and neck region is the Waldeyer's ring, followed by orbit and sinonasal region. 1 Diffuse large B cell lymphoma (DLBCL) is the most common histological type of NHL of the sinonasal region in European and North American patients, whereas sinonasal natural killer (NK)/T cell lymphoma is more common in east Asian and South American patients. 4
ALCL is a rare form of T cell NHL and constitutes 3% of adult and 10%–15% of pediatric and adolescent NHL. 5 It is primarily a nodal disease, but may involve extranodal sites for example, skin, gastrointestinal tract, liver, bone marrow, head and neck, and central nervous system. 6 It is frequently associated with t(2;5)(p23;q35) leading to the creation of a fusion gene consisting of the ALK and the nucleophosmin gene. 7 ALK protein is a transmembrane receptor tyrosine kinase of the insulin receptor superfamily and participates in signaling pathways of the central and peripheral nervous system. 8 As per the current WHO classification, ALCL may be a primary cutaneous or systemic disease and may be ALK positive or ALK negative with 5-year overall survival (OS) rates being 70% versus 49%, respectively (p = 0.016). 9
Sinonasal ALCL is a rare entity. In a review of 120 cases of NHL of the sinonasal tract from the U.S., only one case of ALCL was noted. 10 A brief review of literature pertaining to sinonasal ALCL has been tabulated in Table 1.1,2,11–14 It commonly affects pediatric, adolescent, and young adult patients, mostly of Asian origin.1,2,11–14 The common differentials of this tumor are epithelial malignancies for example, squamous cell carcinoma, adenoid cystic carcinoma, adenocarcinoma, and sinonasal undifferentiated carcinoma; nonepithelial malignancies for example, lymphoma (DLBCL or NK/T cell lymphoma), plasmacytoma, melanoma, and rhabdomyosarcoma; and benign conditions for example, invasive fungal sinusitis, Wegener's granulomatosis, polyp, and inverted papilloma. 1 In view of the initial vague symptoms, difficult access to this anatomical location (for physical examination and biopsy) and myriad diagnostic possibilities, careful examination of the histopathology specimen and ancillary immunohistochemistry study are of paramount importance to prevent diagnostic delay.1,14 The tumor is usually composed of large cells with abundant cytoplasm, round, pleomorphic, reniform, or horseshoe-shaped nuclei with single or multiple prominent nucleoli, giving the cells an epithelial or histiocyte-like appearance.1,2,12–14 The cells strongly express CD30 (Ki-1) and usually express either T cell or null lineage-specific antigens.1,2,12–14 Approximately 60% of cases overexpress the ALK protein.1,2,12–14 In children and young adults, ALCL is usually ALK positive (≥90%). Chromosomal studies, including karyotyping, may be done to demonstrate ALK gene rearrangement, most commonly t(2;5) (p23;q35). 13 In the illustrative case the initial diagnosis at the local referring center was sinonasal MDSCC. Repeat biopsy at our institute suggested the possibility of a hematolymphoid malignancy (pancytokeratin-, CD45+, CD3−, CD20− on immunohistochemistry). Further immunohistochemistry studies (CD4+, CD43+, CD30+, ALK+) finally revealed ALK-positive ALCL.
Sinonasal Anaplastic Large Cell Lymphoma-Review of Literature
F, female; M, male; NC, nasal cavity; LN, lymph node; STM, soft tissue mass; SVCO, superior venacaval obstruction; ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; PD, poorly differentiated; NA, not available; FESS, functional endoscopic sinus surgery; CHOP, cyclophosphamide, Adriamycin (hydroxydaunorubicin), vincristine, and prednisolone; EPOCH, etoposide, prednisolone, vincristine, cyclophosphamide, and Adriamycin (hydroxydaunorubicin); IVAC, Ifosfamide (with Mesna uroprotection), Etoposide, Cytarabine; BMT, bone marrow transplant; BFM, Berlin–Frankfurt–Munster; IFRT, involved field radiotherapy; CR, complete response; FU, follow up; Rx, treatment; PET-CT, positron emission tomography–computed tomography; AIEOP, Associazione Italiana di Ematologia e Oncologia Pediatrica; PR, partial response.
According to NCCN clinical practice guidelines for ALCL in adults, version 4.2018, multimodality management is recommended for the first-line treatment of ALCL in adults. The role of surgery in sinonasal ALCL is limited to an open or endoscopic biopsy for the obtainment of tissue diagnosis. This further emphasizes the critical importance of a prompt and correct diagnosis of this entity to preclude a major surgical resection of a tumor, which is quite sensitive to cytotoxic chemotherapy as well as radiotherapy.11,14 Multiagent chemotherapy with CHOP or CHOEP (CHOP+Etoposide) regimen should be administered for 3–6 cycles depending upon the stage of disease (3–4 cycles in stage I–II and 6 cycles in stage III–IV ALCL). This should be followed by local radiotherapy 30–40 Gray in conventional fractionation. In a study of 46 adult patients with early stage (stage I–20 and stage II–26) systemic ALCL treated with a combination of CHOP or a CHOP-like regimen and IFRT (median dose–46 Gray) the 5-year OS, progression-free survival, and local control rates were 84.4%, 63.6%, and 90.8%, respectively. 3 After a median follow-up of 55 months, no severe late effect or second malignancy was observed in this study. In an earlier Italian multicentric study, 40 adult patients with ALCL–Hodgkin-like subtype were randomized to receive a third-generation high-grade NHL regimen MACOP-B (methotrexate with leucovorin, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin) or a HL-specific regimen ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine). 15 All patients with bulky disease in the mediastinum at diagnosis underwent local radiotherapy (36 Gray) after the completion of chemotherapy. The complete response rates were 90% and 91% and the relapse-free survival rates (at 32 months) were 94% and 91% in the MACOP-B and ABVD arms, respectively. It is notable that 16 out of 19 (84%) patients with mediastinal bulk in this study achieved PR at the end of chemotherapy, and all of these 16 patients subsequently obtained complete response (assessed by serial CT scans and 67Galium SPECT) at the end of mediastinal radiotherapy (RT). However, the use of radiotherapy in the sinonasal region may be associated with acute (fatigue, dermatitis, mucositis, oral ulcers, conjunctivitis, blurring of vision, increased tearing) and late (skin effects, cataract, dimness of vision, xerostomia, dental caries, facial asymmetry in pediatric patients, dysphagia, and second malignancies) adverse effects. Taking into account the anatomical complexity of the disease site and the proximity to critical structures for example, eye, optic pathway, brainstem, and pituitary gland, advanced radiotherapy technique e.g., 3D-CRT or intensity-modulated radiotherapy is strongly advocated. Use of these techniques have significantly decreased the acute and late morbidities of RT, but concern still lingers in the pediatric and adolescent patients. In this age group intensive, multiagent, prolonged, or short-pulse chemotherapy without local RT has been commonly used. In the prospective multicenter NHL-Berlin-Frankfurt-Munster (BFM) 90 trial, 89 patients younger than 18 years of age with newly diagnosed ALCL were stratified into 3 groups (K1: stage I and II resected; K2: stage II nonresected and stage III; and K3: stage IV or multifocal bone disease) and administered a cytoreductive prephase followed by risk-adapted short-pulse B-NHL-type chemotherapy (3 courses in K1; 6 courses in K2; and 6 intensified courses in K3) without local RT. 16 Only one patient received cranial RT (24 Gray) for CNS disease. This approach led to a 5-year event-free survival (EFS) of 76% in the entire cohort (100% in K1; 73% in K2, and 79% in K3). There was no toxic death and one patient had second malignancy (ALL). Two patients had disease progression on chemotherapy and 20 had relapse (18 early relapse and 2 late relapse) after completion of chemotherapy. In the randomized ALCL99 trial conducted by the European Intergroup for Childhood Non-Hodgkin Lymphoma (EICNHL), the NHL-BFM90 backbone was retained and 352 children with standard or high-risk ALCL were randomly assigned to the MTX1 arm (n = 175; injection methotrexate 1 g/m2 over 24 h and intrathecal chemotherapy) and the MTX3 arm (n = 177; injection methotrexate 3 g/m2 over 3 h without intrathecal chemotherapy). 17 There was no significant difference in the 2-year EFS rates of 73.6% and 74.5%, and the 2-year OS rates of 90.1% and 94.9% in the MTX1 and MTX3 arms, respectively. However, the incidence of grade 4 hematologic toxicity, infection, and grade 3–4 stomatitis was significantly higher in the MTX1 arm. In this study, four patients had toxic death as the first event. Early disease progression and relapse (87% early relapse) were noted in 14 and 84 patients, respectively. Addition of vinblastine during induction and as maintenance for a total treatment duration of 1 year in high-risk patients with ALCL (mediastinal, lung, liver, spleen, or skin involvement), enrolled on this trial, significantly delayed the occurrence of relapses but did not reduce the risk of failure. 18 In the CCG-5941 trial, 86 children with nonlocalized ALCL were treated with a compressed aggressive multiagent T-cell lineage chemotherapy regimen consisting of a 3-week induction therapy (vincristine, prednisone, cyclophosphamide, daunomycin, asparaginase) followed by a 3-week consolidation period (vincristine, prednisone, etoposide, 6-thioguanine, cytarabine, asparaginase, methotrexate) followed by six courses of maintenance chemotherapy at 7-week intervals (cyclophosphamide, 6-thioguanine, vincristine, prednisone, doxorubicin, asparaginase, methotrexate, etoposide, cytarabine) without local RT leading to a 5-year EFS of 68% and a 5-year OS of 80%. Only 1 patient with CNS disease at diagnosis received cranial RT (18 Gray) at the end of therapy. There were 21 relapses (81% early relapse) and 4 toxic deaths (due to infection) as first events. Hematological toxicities were profound (grade 4 neutropenia 82%, grade 4 thrombocytopenia 66%, and grade 4 anemia38%) and 4% patients developed grade 4 hepatoxicity. 19 These studies portray the fact that intensive multiagent chemotherapy without local RT leads to impressive EFS of 65%–75% and OS of 70%–90% at 5 years in pediatric patients with ALCL. However, roughly 1/3rd (20%–40%) of these patients experience early relapse, mostly at the site of initial involvement. Around 80% of these patients are successfully salvaged with further multiagent chemotherapy, immunotherapy (brentuximab vedotin), and biological therapy (crizotinib, ceritinib). The acute (severe hematological toxicities, neutropenic fever, infection, stomatitis, hepatoxicity, and toxic death up to 5%) and late (cardiomyopathy and congestive cardiac failure, infertility, and second malignancy) toxicities of such intensive multiagent chemotherapy regimen should be carefully considered in this age group. The illustrative patient had stage IE ALK-positive ALCL and was treated with a combination of 4 cycles of chemotherapy with CHOP regimen followed by local radiotherapy 36 Gray in 20 fractions over 4 weeks by 3D-CRT technique. The expected 5-year OS rate in this clinical scenario is in the range of 70%–80% and our patient is alive and disease free for 40 months from the initial diagnosis. Long-term follow-up is required to detect late recurrence and late effects of treatment.
Conclusion
Sinonasal ALK-positive ALCL is a very rare tumor. It has been predominantly reported in pediatric, adolescent, and young adult patients, mostly of Asian origin. Awareness of this rare entity, meticulous examination of the histopathology specimen, and relevant immunohistochemistry tests are crucial to avoid any diagnostic dilemma. Abbreviated chemotherapy with 4 cycles of CHOP regimen followed by local conformal radiotherapy with a modest dose (36 Gray in conventional fractionation) is a reasonable treatment option for early stage sinonasal ALK-positive ALCL in adults.
Footnotes
Acknowledgments
This work received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. The patient gave written informed consent before the administration of chemotherapy and radiotherapy. Informed consent for patient information and images to be published was also provided by the patient.
Author Disclosure Statement
No competing financial interests exist.