
Abstract
Juvenile granulosa cell tumor (JGCT) of the ovary is an uncommon malignancy, with most cases seen in adolescent girls and young women. The majority of these patients present with unilateral ovarian disease, and to date, bilateral JGCTs have been reported in 10 cases. Although the histopathologic features have been detailed in the published literature, extensive extracellular mucin deposition has been documented in only one case. Herein, we report a 17-year-old adolescent girl with bilateral solid-cystic adnexal masses diagnosed as bilateral JGCT with abundant extracellular mucin deposition on histopathology. The index case highlights a rare clinical and histopathologic presentation of JGCT. Adequate knowledge of such unusual presentations is essential for accurate distinction from other ovarian tumors and appropriate management.
Introduction
Granulosa cell tumors (GCT) comprise around 1%–5% of all ovarian tumors. Adult GCTs (AGCT) are more common and account for 95% of all GCTs, while juvenile GCTs (JGCT) account for around 5% of cases. 1 JGCTs commonly occur in girls and young women <30 years of age, with most cases showing unilateral involvement. Bilateral ovarian involvement in JGCTs is unusual and has been reported in 2% of these cases. 2 The clinical presentations are often nonspecific and similar to those seen in other ovarian tumors; however, a majority of pre-menarchial girls present with isosexual precocious puberty due to excess hormone secretion by the neoplastic granulosa cells.1,2
Histopathologic examination of the tumor is essential for a definitive diagnosis. However, establishing a histopathologic diagnosis based on the morphology alone can be challenging as the pathologic features may overlap with various other ovarian malignancies.2–4 This diagnostic difficulty is compounded by the rarity of the tumor and the lack of awareness of its pathologic and immunohistochemical features among surgical histopathologists.
A variety of histomorphological patterns have been described in JGCTs in the literature. The most common architectural patterns include the macrofollicular and solid arrangement of tumor cells.2,4,5 A limited number of reports in the literature have documented the presence of significant nuclear pleomorphism, microcystic, and pseudopapillary patterns with hob nailing, extensive sclerosis, and the presence of hyaline bodies.2–4 Variable amounts of eosinophilic or basophilic secretions have been described in the lumen of the microfollicles and macrofollicles; however, the presence of widespread extracellular mucin deposition in the tumor stroma in ovarian JGCTs has been reported infrequently in only one of the previously published JGCT cases. 6 Owing to such rarity, its association with adverse/favorable clinical course in these patients has not been studied.
Herein, we present a case of a JGCT in an adolescent with bilateral ovarian involvement and abundant extracellular mucin deposition.
Case Report
A 17-year-old adolescent girl presented with gradually progressive abdominal distension and dull aching pain for the last 3 months. She had attained menarche at 13 years, and her menstrual cycles were regular. She denied any history of fever, nausea, vomiting, jaundice, constipation, hematuria, and weight loss or loss of appetite. On examination, there was abdominal distension up to the umbilicus; however, there was no free fluid. Ultrasonography of the abdomen and pelvis revealed bilateral solid-cystic adnexal masses measuring 9 × 8 and 8 × 8 cm. Cystic areas were variable-sized, multiloculated with thin septae; however, they did not show papillary projections. The uterus, cervix, parametria, and omentum were unremarkable.
No free fluid was detected in the pouch of Douglas, and there was no significant abdominopelvic lymphadenopathy. Her serum CA-125 was elevated (560 IU/mL). Based on these clinicoradiologic features, a provisional diagnosis of ovarian malignancy was made. Ultrasound-guided fine-needle aspiration cytology attempted at a peripheral medical center suggested a cytologic diagnosis of epithelial ovarian malignancy. Subsequently, the patient underwent bilateral salpingo-oophorectomy and infracolic omentectomy with preservation of the uterus.
Formalin-fixed paraffin-embedded tissue blocks were submitted for a histopathologic review at our institute. On review, sections from these blocks showed a malignant tumor with variable architectural patterns. The tumor was arranged in sheets, nests, cords, trabeculae, and microcystic and macrocystic areas with cysts filled with predominantly basophilic and eosinophilic secretions. Large pools of abundant pale-staining extracellular mucinous material with clusters, papillae-like arrangements, and cords of tumor cells floating in the mucin lakes were noted. Focally, solid nests with microfollicular arrangement were also noted. The individual tumor cells exhibited mild to moderate nuclear pleomorphism with round to oval nuclei, fine chromatin, small nucleoli, occasional nuclear grooves, and a moderate amount of pale to eosinophilic cytoplasm.
In addition, many loose clusters and singly scattered luteinized cells were noted. Mitosis was 3–4/10 high-power fields (HPFs). However, there were no atypical mitoses, marked nuclear atypia, or areas of necrosis (Figs. 1 and 2). A histopathologic diagnosis of a malignant ovarian tumor, with differential diagnoses of JGCT and yolk sac tumor, was rendered based on the clinical details and microscopic features.
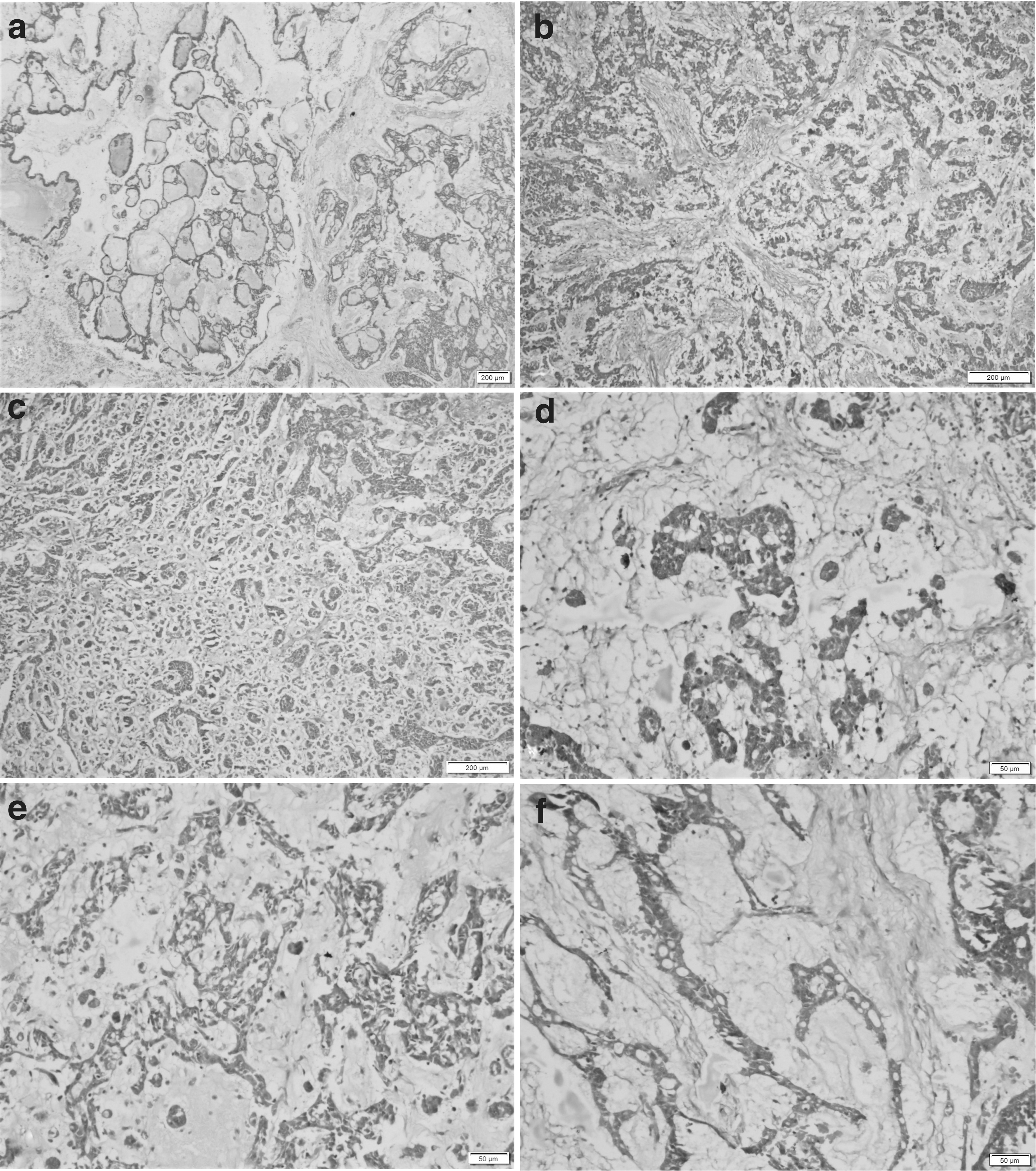
Panel of photomicrographs showing the extensive mucin deposition in bilateral ovarian juvenile granulosa cell tumor.

Panel of photomicrographs showing the other areas in the ovarian juvenile granulosa cell tumor.
Immunohistochemistry was performed to confirm the diagnosis. The tumor cells showed diffuse nucleocytoplasmic positivity for calretinin and cytoplasmic positivity for inhibin (confirming a sex-cord origin). They were negative for SALL4, glypican 3 (excluding yolk sac tumor), and pan-cytokeratin (excluding a mucinous and clear cell carcinoma) (Fig. 3). Thus, a final diagnosis of bilateral ovarian JGCT with extensive extracellular mucin deposition was rendered in this patient. The patient was planned for post-operative adjuvant chemotherapy (six cycles of bleomycin, etoposide, and cisplatin regimen). The decision for post-operative adjuvant chemotherapy was taken because of bilateral ovarian involvement, and the details of any surgical spill or capsule details being unknown. However, unfortunately, probably due to the current pandemic, the patient was lost to follow-up.
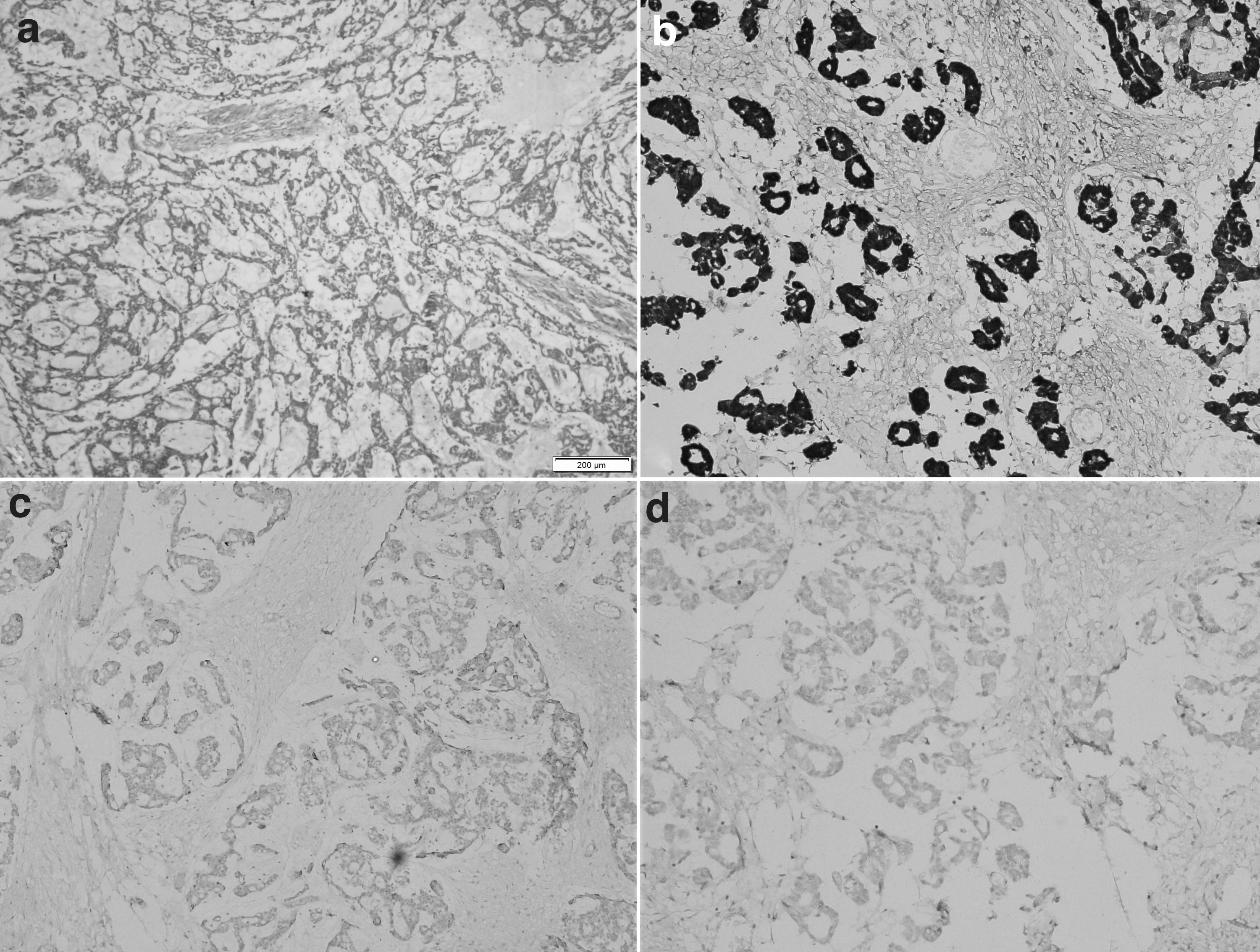
Discussion
Ovarian neoplasms are rare in children and adolescents and constitute around 1% of all tumors occurring in this age group. Among the ovarian neoplasms seen in children and adolescents, germ cell tumors are the most common, constituting around 70% of all ovarian tumors in the pre-menarchial age group. 7 Granulosa cell tumors are much less frequent and comprise around 1%–3% of all ovarian tumors. 2
There are two types of granulosa cell tumors, adult (AGCT) and JGCT. These types are distinct entities, which differ in their incidence, age at presentation, etiopathogenesis, and histopathology. The FOXL2 (C402G) somatic mutation is consistently observed in around 97% of AGCT cases and hence is considered the molecular hallmark of AGCTs. 8 In contrast, to date, no such genetic alteration has been identified in JGCTs, and its etiopathogenesis is largely unknown. However, a few studies have reported the occurrence of AKT and gsp mutations in JGCT cases. 9
Furthermore, unlike AGCT, JGCT accounts for only 5% of all GCTs, with most patients presenting in the first two decades of life with either nonspecific manifestations like abdominal distension and pain or with isosexual precocious puberty due to hormone secretion.2,10 The index patient presented at 17 years of age with gradually progressive abdominal distension and pain.
The JGCTs are unilateral in a vast majority (95%–98%) of patients with variable radiological appearances, most common being solid-cystic masses, followed by purely cystic masses without papillary projections, with purely solid masses being the least typical radiologic appearance.2,11 In the index patient, the radiologic investigations revealed bilateral solid-cystic ovarian masses. To estimate the approximate frequency of bilateral JGCTs, we performed a comprehensive nonsystematic literature review from the year 1940 till 2021. We found that bilateral ovarian involvement in JGCTs is extremely rare, with mere ten such cases documented in the literature.
Table 1 shows the clinical presentations, pathological features, management, and follow-up details of all the bilateral JGCT cases documented in the literature to date.2,3,12–17 In addition to these ten cases, two cases in the literature developed contralateral JGCT within one year and 10.5 years of the surgical removal of the initial unliteral tumors (possibly metachronous or metastatic).2,17
Review of the Published Literature, Enumerating Cases of Ovarian Juvenile Granulosa Cell Tumors, with Bilateral Ovarian Involvement
AGCT, adult granulosa cell tumor; BEP, bleomycin, etoposide, cisplatin; HPFs, high-power fields; IHC, immunohistochemistry; LVSI, lymphovascular space involvement.
On microscopy, JGCT can demonstrate a variety of architectural patterns, with solid, macrofollicular, and microfollicular patterns being the most frequently noted ones. The tumor cells are round to polygonal, with round to oval nuclei having moderate to marked pleomorphism, fine to coarse chromatin, prominent nucleoli, and a moderate amount of pale-vacuolated to luteinized cytoplasm. Frequent mitotic figures are noted. In general, its microscopic appearance is worse compared to its actual prognosis.2–4
A few studies in the literature have reported some unusual histomorphologic patterns.2–4,6 In the most extensive JGCT series to date, Young et al. studied a total of 125 JGCT cases. In addition to the common histopathological findings, they reported a focal tubular pattern in 6 (4.8%) of their cases, a random admixture of granulosa and theca cells in 44 (35.2%), sclerosing stromal tumor-like morphology in 14 (11.2%), foci of calcification in 2 (1.6%), intercellular edema in 10 (8%), intracellular and extracellular hyaline bodies in 3 (2.4%), and areas of hemorrhage and/or necrosis in 43 (34.4%) of their cases. 2
In a recently reported series of seven JGCT cases, the authors observed reticular and microcystic architecture resembling yolk sac tumor in two (28.6%) cases, and myxoid stroma intermixed with follicular fluid and sarcomatous appearance in one (14.3%) case each. 4 In another recent series of 15 JGCT cases, 1 (6.7%) case demonstrated tubulocystic areas with marked nuclear pleomorphism and hob nailing, closely resembling yolk sac tumor. Three (20%) cases showed extensive stromal hyalinization, and multiple thick hyalinized blood vessels and focal areas of myxoid degeneration were noted in one (6.7%) case each. 3 A single report documents a recurrent JGCT in a 34-year-old woman with abundant extracellular mucin that led to an initial misdiagnosis of the tumor as a mucinous adenocarcinoma.
The authors highlight the utility of an appropriate immunohistochemical panel, including inhibin and/or calretinin, for a definitive diagnosis in such cases. 6 Similar pools of extracellular mucin with small clusters of neoplastic granulosa cells floating were noted in the index case.
Owing to its histomorphologic diversity, JGCT often poses a significant diagnostic challenge. The diagnostic difficulty is compounded when unusual microscopic features are present. The bilateral ovarian tumors demonstrated extensive extracellular mucin deposition in the index patient with sheets, cords, and nests of mildly to moderately pleomorphic tumor cells. In adolescents and young adults presenting with bilateral ovarian tumors with abundant extracellular mucin, a variety of differential diagnoses, including yolk sac tumor, JGCT and rare mucinous, and clear cell adenocarcinoma, need to be considered. Table 2 highlights the characteristic clinicopathologic features that can help in differentiating such JGCTs from the common histopathologic mimics.
Distinctive Clinicopathologic Features of Histopathologic Mimics of Ovarian Juvenile Granulosa Cell Tumor with Extensive Extracellular Mucin Deposition
Other significant histopathologic differentials of JGCT are AGCT, small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), small cell carcinoma of the ovary, pulmonary type (SCCOPT), thecoma, ovarian large cell neuroendocrine carcinoma, poorly differentiated carcinoma, ovarian non-Hodgkin's lymphoma, endometrial stromal sarcoma, and rarely, yolk sac tumor.3,18–20 Extensive sampling, thorough microscopic examination, and an appropriate immunohistochemical panel can help in reliably distinguishing these tumors. 21
Although rarely, AGCTs can occur in young patients, may be associated with symptoms due to androgen production, and may show focal areas with higher nuclear pleomorphism, and hence may mimic the JGCTs. Similarly, rare JGCTs can be seen in older women. Subtle morphologic cues favoring a diagnosis of JGCT over AGCT in such cases include a predominance of irregular sized and shaped follicles, a higher degree of nuclear pleomorphism, brisk mitosis, numerous luteinized cells, lack of longitudinal nuclear grooves, and Call-Exner bodies. Demonstration of the presence of FOXL2 point mutation in AGCT cases (and its absence in JGCT) is diagnostic. 8 Immunostaining for FOXL2 protein, however, shows nuclear positivity in both AGCT and JGCT, irrespective of the FOXL2 mutation status. 3
The predominant tumor cells in SCCOHT, are small and primitive-appearing, and can demonstrate focal follicular architecture, thus mimicking JGCT. However, this distinction is imperative as SCCOHT has a much aggressive course, worse prognosis, and poorer clinical outcome than JGCT. 22 Immunohistochemistry for inhibin, SMARCA4/BRG, epithelial membrane antigen, and INI1 can reliably help in confirming the diagnosis. 21 The presence of abundant extracellular mucin may lead to a misinterpretation of these tumors as mucinous adenocarcinoma. However, careful clinical, pathologic, and immunohistochemical correlation can help differentiate it from a JGCT. 21
JGCT carries a good prognosis, especially when diagnosed in the early stages, reiterating that the tumor stage is the most important prognostic factor. Some studies report an increased risk of recurrence with moderate to severe nuclear pleomorphism, frequent mitosis (>4/10HPF), residual disease post-surgery, and FOXL2 immunoexpression.2,16
Surgery is the mainstay of treatment, with an evolving role of chemotherapy. Surgery alone has been advised for cases diagnosed in stage I. For young women, fertility-preserving surgeries are now being advocated, especially for those who wish to bear children in the future.1,23–25 Fertility-preserving surgeries include cystectomy or ipsilateral oophorectomy with/without omentectomy and/or lymphadenectomy.23–25 To date, there are no standardized treatment protocols regarding the use of adjuvant chemotherapy in JGCT, owing to its rarity.
However, most commonly, post-operative chemotherapy is used for all JGCT patients with disease beyond FIGO stage Ic and those with recurrent/residual/unresectable disease. The most common chemotherapeutic regimen uses a combination of bleomycin, etoposide, and cisplatin. The index patient was also planned for post-operative chemotherapy; however, she was lost to follow-up. Radiotherapy may be employed for the management of residual/recurrent disease. 16
Conclusions
Juvenile GCT is a rare ovarian malignancy that can exhibit a variety of histomorphological patterns. This report documents an extremely rare bilateral ovarian JGCT with extensive extracellular mucin deposition that has been described in only one case previously. Adequate knowledge of its characteristic and unusual histopathologic and immunohistochemical features is essential for accurate diagnosis and appropriate management in such cases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this study.