
Abstract
The cDNA sequence of feline interferon receptor 2 (feIFNAR2) was generated using RT-PCR method in present study. This gene included 1,572 bp and encoded a 523 aminoacid (aa) protein with a 35 aa signal peptide. The deduced protein shared 61% amino acid identity to the human IFNAR2. There were two fibronectin type III (FBN-III) domains of about 110 residues in the extracellular domain. Homology modeling of feIFNAR2 presented a similar structure with other IFN receptors. The ELISA and FACS experiments demonstrated that the protein could bind to feIFN-α or feIFN-ω. However, antiviral activity assay found that feIFN-ω had broader species spectrum compared with feIFN-α. To define the functional differences, several point mutations of feIFNAR2 were constructed and the relative affinities of feIFN-α or feIFN-ω for feIFNAR2 and mutants were evaluated. The results suggested that feIFN-α and feIFN-ω had different binding sites on feIFNAR2. T75 and M77 on feIFNAR2 were hotspots for binding to feIFN-α, but not to feIFN-ω. These findings suggested that the cloned feline IFNAR2 interacted with both feIFN-α and feIFN-ω, however, not sharing the same binding sites.
Introduction
T
A functional type I receptor consists of two subunits, IFNAR1 and IFNAR2, both of which are in the class II cytokine receptor family (reviewed by Walter 2004). IFNAR1, alone, generally appears not to bind type I IFN in detectable level (Cohen and others 1995), and a second subunit (IFNAR2) is necessary to allow IFN to bind with high affinity to the receptor (Uzé and others 1990; Uzé and others 1992). The soluble form of human IFNAR2 was identified in 1994 (Novick and others 1994). The structure of the extracellular domain (EC) of human IFNAR2 has been solved using NMR (PDB id 1N6U) (Chill and others 2003). Human IFNAR2-EC is comprised of two fibronectin type III (FBN-III) modules, which are characterized by seven β-strands arranged in a mutual perpendicular β-sandwich (Langer and others 2004). The IFN-binding site on IFNAR2 is located on loops 43–53 and 76–80, and the interdomain residues 100–110 (Chuntharapai and others 1999; Roisman and others 2001; Chill and others 2003). Although type I IFNs bind to the same region, they have distinct binding sites on IFNAR2. M46 is a specific hotspot for IFN-α, while W100 is a specific hotspot for IFN-β binding. In cat, many IFNs have been discovered (Nakamura and others 1992; Wonderling and others 2002; Yang and others 2007), and they differ in their inducibility and hence may provide a flexible response to infection by different pathogens (Minagawa and others 1999; Martin and others 2002; de Mari and others 2003, 2004). In this study, to understand the difference in antiviral activity between feIFN-α and feIFN-ω, the cDNA of feline IFNAR2 was isolated and the receptor-binding capacity between feIFN-α and feIFN-ω was compared. The present data showed that the distinct binding sites for feIFN-α and feIFN-ω on feline interferon receptor 2 (feIFNAR2), residues T75 and M77, were important for feIFN-α binding, but not critical for IFN-ω. Using crystal structures from human IFNAR2 extracellular domain (IFNAR2-EC) as template, the most probable structure of feIFNAR2-EC was presented by 3D homology modeling.
Materials and Methods
RNA isolation and RT-PCR
The spleen from a short-haired cat was collected, and its total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA). Then the total RNA was reverse-transcribed using 200 U Superscript™ II Reverse Transcriptase (Invitrogen, Carlsbad, CA), then the cDNA was used as a template for amplifying feIFNAR2. The special primers were designed; the forward primer 5′-tgt atct cgt ggt gtg tat-3′, the reverse primer 5′-tga ttc aga act gtc act t-3′. The amplification conditions were as follows: 94°C for 10 min, followed by five cycles of 94°C for 1 min, 45°C for 1 min, and 72°C for 1 min, 10 cycles of 94°C for 1 min, 50°C for 1 min, and 72°C for 1 min, 15 cycles for 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. The final extension was at 72°C for 10 min. The amplified product of ∼1,500 bp was purified from agarose gel. The purified PCR product was cloned into the pMD18-T vector (Takara, Kyoto, Japan) and then sequencing. The recombinant T-vector was named pT-feIFNAR2. Then the whole sequence was obtained by alignment with feline genome.
Cell transfection with PEI
Polyethylenimine (PEI) was obtained from Sigma-Aldrich (No. G8416). 293T Cells were seeded a day prior to transfection in 10-cm dish at a density of 106 cells. One to two hours prior to transfection remove media and replace with 2 mL DMEM with 10% FBS. Six micrograms of plasmid and 18 µL of PEI (from 2 mg/mL solution) were diluted with 0.9% NaCl, respectively. After incubating for 8 min at room temperature, they were mixed and were incubated for 20 min at room temperature. Then the mixture was added to cells. After 12 h, cells were changed medium with 10 mL DMEM with 10% FBS. Then cells were grown for 36 h at 37°C with 5% CO2.
Binding assay for feIFNAR2 and IFN in 293T cells
The extracellular domain and transmembrane region (1–273 aa) of feIFNAR2 was amplified by PCR from the pT-feIFNAR2 plasmid using primers that incorporated ApaI and KpnI restriction sites. Amplified fragments were cloned into a pCMV-myc vector (Clontech, Mountain View, CA). 293T cells transiently transfected with the recombinant plasmid using PEI. After 48 h, the cells were harvested and washed with PBS (3% BSA) for three times, then incubated with feIFN-α or feIFN-ω (5 nM) on ice for 1 h. After being washed three times, the cells were incubated with rabbit anti-feIFN-ω antibody for 1 h, then adding FITC-labeling anti-rabbit antiserum for 1 h. After being washed three times, the cells were suspended in fluorescence-assisted cell sorting (FACS) buffer (PBS plus 3% BSA). FACS was performed with BD FACSAria. About 10,000 cells were analyzed for each sample. The acquired data were analyzed with FlowJo (Tree Star, Inc., USA) software.
Expression and purification of feIFNAR2-EC from Escherichia coli
The feIFNAR2-EC (35–252 aa) fragment was generated with feIFNAR2-EC-F (5′-gcc ata tgt ggc ctg att tgt cag a-3′) and feIFNAR2-EC-R (5′-gac tcg agt ttg gca gat tct gat g-3′) as primers and cloned into the prokaryotic expression vector, pET-30a (Novagen, Darmstadt, Germany), through the restriction sites (NdeI and XhoI) introduced by PCR. The recombinant vector was named pET-30a-feIFNAR2-EC. For protein production, the cells containing the pET-30a-feIFNAR2-EC plasmid were grown at 37°C in 1 L of LB medium containing 100 µg/mL kanamycin. At A 600 = 0.6, protein expression was induced by the addition of 0.2 mM IPTG. After 10 h of additional growth at 16°C, the cells were harvested and treated under sonication, the insoluble fraction was separated by centrifugation, and the inclusion bodies were washed with PBST (1% triton), 2 M urea, and 1 M NaCl by turns. Then the inclusion bodies were dissolved in 8 M urea. The recombinant proteins were analyzed on 12% SDS-PAGE. Then the fused protein was refolded by a 20-fold dilution in the refolding buffer: 50 mM Tris, pH 8.0, 0.5 M Arg, 2 mM EDTA, 0.5 mM glutathione (oxidized), and 20% glycerol. After 48 h in 4°C, the solution was centrifuged at 12,000 rpm and the precipitation was discarded. Finally, the solution was concentrated by dialysis in the refolding buffer, without glycerol. Protein concentrations were determined from the absorbance at 595 nm using Bradford method.
Site-directed mutagenesis
Site-directed mutagenesis was carried out by PCR amplification of the expression plasmid pET-30a with 18–21 nucleotide primers containing the mutated codon using TaKaRa Pyrobest DNA polymerases (TaKaRa, Kyoto, Japan). After restriction digestion and ligation, the mutated plasmids were transformed into E. coli Rossetta (DE3) (Novagen, Darmstadt, Germany) cells. The sequences of the whole expressed gene containing the mutations were verified by DNA sequencing. The expression, purification, and refolding were processed as the wild feIFNAR2-EC.
ELISA for the binding of IFN to feIFNAR2-EC
One microgram IFN-α or IFN-ω was added into the wells that had been coated with the purified recombinant feIFNAR2-EC or mutants (500 ng/well) overnight at 4°C. After 1 h of incubation at 37°C and six washes with PBST (0.2% Tween), the wells were incubated with rabbit anti-feIFN-ω antibody (1:100,000) for 1 h at 37°C and then incubated with HRP-labeled secondary antibody (1:10,000) for 1 h at 37°C. The color was developed using mixture of TMB (tetramethyl benzidine) and H2O2 (hydrogen peroxide) solution and the reaction was stopped using 50 µL of 1 M H2SO4. Absorbance values were measured in an ELISA plate reader at a wavelength of 450 nm.
Antiviral protection activity assay
Antiviral activities of feIFN-α and feIFN-ω were determined by the inhibition of the cytopathic effect of virus on cells as described (Wonderling and others 2002). The activities of IFNs were determined from the relative concentration needed for 50% protection of the cells compared to Intercat (Toray Industries, Tokyo, Japan).
Homology modeling of IFNAR2-EC
A structure model of the extracellular part of the IFNAR2 (218 aa) from WPDLS (N-ter) to ESAK (C-ter) was built using homology modeling. The structure of human IFNAR2-EC (PDB id 1N6U) (Chill and others 2003) was used as template for homology modeling with SWISS-MODEL (Guex and Peitsch 1997; Schwede and others 2003; Arnold and others 2006). The resulting theoretical models were displayed and analyzed using DeepView/Swiss-Pdb Viewer 4.0 program (http://www.expasy.org/spdbv/).
Results
Isolation and characterization of the feline IFNAR2
In this study, the cDNA of the feIFNAR2 (1,572 bp) was isolated by RT-PCR from feline spleen. The primers for amplifying the feIFNAR2 were designed according to the known IFNAR2 sequences, including human, bovine, sheep, and canine IFNAR2. The PCR product was 1,481 bp in length and the sequence was aligned with feline genome sequence in GenBank. Finally, the whole sequences of feIFNAR2 were obtained (Fig. 1). The predicted amino acid sequences of feIFNAR2 contained 523 amino acid residues. It included a signal sequence (1–34 aa) and a mature protein using the soft signalP 3.0 (Bendtsen and others 2004). The mature protein had three parts, composing of extracellular domain of 218 amino acids, transmembrane domain of 21 amino acids, and intracellular domain as the arrangement of other IFNAR2. The extracellular domain was made of two FBN-III domains with Ig-like folding topology.

DNA and predicted amino acid sequences of feline IFNAR2. Conserved cysteine residues are circled. The predicted signal peptide and transmembrane domains are underlined. The boundaries of the transmembrane domain are predicted by alignment with huIFNAR2. The potential N-linked glycosylation sites are underlined with broken lines. The conserved proline and box 1 motif are boxed.
Alignment of the predicted amino acid sequences of the feline IFNAR2 with those of dog, human, cow, and sheep is shown in Figure 2. As expected, there is extensive sequence identity between fe- and caIFNAR2 (73% identity) reflecting the close evolutionary relationship of these two species. The deduced amino acid sequence of feIFNAR2 showed 61%, 61%, and 59% identity with human, bovine, and sheep counterparts, respectively (Table 1). The placement of the six extracellular cysteine residues was identical in fe-, ca-, hu-, bo-, and ovIFNAR2. The extracellular domain of those proteins had three common potential N-glycosylation sites. A proline-rich box 1 motif and the C-terminal sequence in the membrane proximal region of cytoplasmic domain were highly conserved for IFNAR2, which had been interpreted to be important in activating the Jak-Stat pathway and triggering an antiviral state after human IFN-α2 binding to the IFNAR2 (Domanski and others 1997). Two tyrosine sites in GYTM and GYIM motif may play an important role in the STAT activation (Zhao and others 2008).
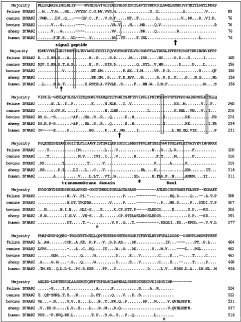
Alignment of feIFNAR2, caIFNAR2, boIFNAR2, ovIFNAR2, and huIFNAR2. Consensus sequence is shown above five sequences. Signal peptides may be as long as that in human proteins. Four conserved cysteine residues in each extracellular FBN-III domain are numbered. Potential sites for N-glycosylation are labeled (↑). A proline-rich box 1 motif and a proline (#) are marked, which are potentially important in Jakase binding. Two conserved tyrosine (*) may play an important role in Stat activation. Signal peptides and transmembrane domain are boxed.
Homology structure of IFNAR2-EC
The alignments used for model calculation showed 62% amino acid similarity between feIFNAR2-EC and huIFNAR2-EC. 3D models of feIFNAR2-EC were generated by using SWISS-MODEL service (http://swissmodel.expasy.org) and adjusted by hand based on the published data of the monomer of human IFNAR2-EC. The structure of feIFNAR2-EC was very similar to that of huIFNAR2-EC (Fig. 3). Both of them folded into two FBN-III domains, which were linked by 10 residues interdomain (V131 to I140). There were six cysteine residues as that in the huIFNAR2-EC, four of which was conserved in the class II cytokine receptor family (reviewed by Walter 2004). There were five N-glycosylation sites, which of two were specific in feIFNAR2-EC. As the structure of human IFNAR2-EC, residues of T75, V76, M77 were in the surface of the protein.
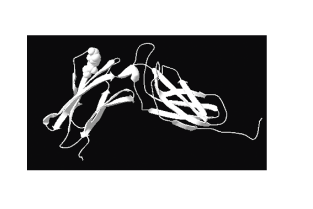
Homology modeling of the extracellular domain of feIFNAR2-EC. The structure was analyzed and all pictures were created by the SWISS-MODEL program and DeepView/Swiss-Pdb Viewer 4.0 program. Residues of T75, V76, and M77 were indicated as spheres.
IFN binding to feIFNAR2 expressed on 293T cells
The assay of antibody titration against feIFN-α and feIFN-ω was performed for the following binding assay that evaluates the interaction between IFNs and felFNAR2. The ELISA data indicated there were no differences between the binding abilities of the antibody to feIFN-α and feIFN-ω at higher concentration (Fig. 4A). To detect whether the cloned protein bind to feIFN-α and feIFN-ω, the 293T cells were transiently transfected with the plasmid of pCMV-myc-feIFNAR2 (1–273 aa). Western blot assay showed that the recombinant protein was expressed in the 293T cells, and the analysis of co-localization with fluorescence membrane marker YFP-MEM indicated that the feIFNAR2 (1–273 aa) could insert into the membrane (data not shown). Then the cells expressed feIFNAR2 (1–273 aa) were incubated with feIFN-α or feIFN-ω (5 nM) on ice for 1 h. After being washed three times, the cells were incubated with rabbit anti-feIFN-ω antiserum for 1 h, then added FITC-labeling anti-rabbit antiserum for 1 h. After being washed three times, the cells were suspended in FACS buffer (PBS plus 3% BSA), and analyzed with BD FACSAria. The results showed that both feIFN-α and feIFN-ω could bind to the feIFNAR2 (Fig. 4B), but the relative affinity had weakly different. The binding strength between feIFN-ω and IFNAR2 was higher than that between feIFN-α and IFNAR2.
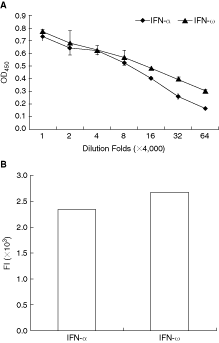
Feline interferon-α (FeIFN-α) and feIFN-ω bind to feIFNAR2 using FACS. (
Comparison of feIFN-α and feIFN-ω binding to feIFNAR2-EC
In order to know the difference between feIFN-α and feIFN-ω binding to feIFNAR2, the extracellular domain of feIFNAR2 (feIFNAR2-EC) and mutants were expressed in Rossetta (DE3). The expressed protein was 24 kDa, as the expected size. All feIFNAR2-EC proteins precipitated in the form of inclusion bodies (Fig. 5, lane 3), which were solubilized in 8 M urea. Refolding was initiated by a 20-fold dilution in the refolding buffer. SDS-PAGE analysis under non-reducing conditions (Fig. 5, lane 4) revealed that <50% of the protein was monomeric, while the rest formed multimeric, disulfide-linked aggregates. Purification of mutants was processed as that of feIFNAR2-EC.

Expression and purification of feline IFNAR2-EC in Escherichia coli. Cells harvested overnight after induction of protein expression at 16°C (lane 1). The cell content was fractionated to supernatant (lane 2) and precipitation. Lane 3 showed the protein expressed by the form inclusion body (IB). The IB was washed by PBST (1% Triton), 2 M urea, and 1 M NaCl, and was solubilized in 8 M urea. The results indicated that the purified protein only partly was monomer (lane 5).
The region presumably involved in the interaction with IFN could be identified by the structures of the complex of human IFNAR2-EC/IFN (Chill and others 2003). Functional contributions of some residues in this region have been confirmed by a mutational study on IFNAR2 (Lewerenz and others 1998; Piehler and Schreiber, 1999b). To find the important binding sites for IFN on feIFNAR2, five mutations were constructed: T75A, T75A/D82N, V76A, M77A, and M77I. The relative affinity to feIFN-α or feIFN-ω was investigated by ELISA (Fig. 6). The binding affinities of the mutant proteins toward feIFN-α were different to wild-type IFNAR2-EC. The residues T75 and M77 were identified as hotspots for binding of feIFN-α, decreasing binding by >50%. The mutant of M77I greatly decreased the binding ability to IFN-α rather than IFN-ω. The mutant of V76A did not change the binding to feIFN-α. Besides, the single FBN-III fragments of IFNAR2-EC also decreased the affinity with feIFN-α. However, all mutants of IFNAR2-EC were not critical to the binding to feIFN-ω. The results indicated that the feIFN-α and feIFN-ω do not share the same binding sites on the IFNAR2, which were coupled with the different activities of feIFN-α and feIFN-ω.
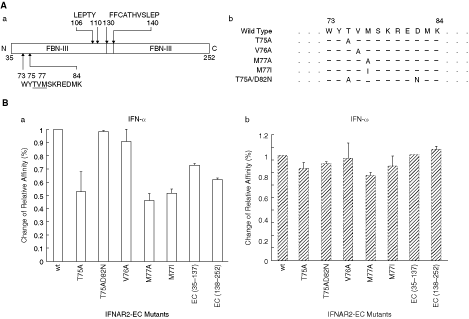
Comparison of residues in the binding sites of IFNAR2 to binding feline interferon-α (feIFN-α) and feIFN-ω. (
Antiviral activities of feIFN-α and feIFN-ω on cross-species cells
To verify the mutant experiment, the antiviral activities of feIFN-α and feIFN-ω on cross-species cells against vesicular stomatitis virus (VSV) were tested. Table 2 showed the antiviral activities among feIFN-α and feIFN-ω on cat kidney (CRFK) cells, bovine kidney (MDBK) cells, porcine kidney (PK-15) cells, human cervix carcinoma HeLa cells, and African green monkey kidney (Vero) cells. These two IFNs had high antiviral activity on CRFK cells. The antiviral activity of feIFN-ω was about double that of feIFN-α in CRFK cells. Comparison to antiviral activity in deferent types of cells, feIFN-α and feIFN-ω had measurable antiviral activity on the MDBK, PK-15, HeLa, and Vero cells, but the antiviral activity of feIFN-α was significantly lower than that of feIFN-ω. Therefore, of all IFN tested, feIFN-ω showed relatively broad cross-species activity; however, feIFN-α had antiviral activity only on the feline cell line. The data were accord with the mutant experiment, which suggested that feIFN-ω and feIFN-α did not share the same binding sites on feIFNAR2.
aThe titer of the antiviral activity, ×106 U/mg.
bAntiviral activity of Intercat IFN is shown as units per viral.
Antiviral activity of feIFN-α and feIFN-ω was assayed by the inhibition of the cytopathic effect of cells challenging with virus. The units of activity (U/mg) were calculated by the Reed-Muench method. The antiviral activity was relative to the Intercat. Displayed values are the mean of experiments carried out in triplicate.
Discussion
We cloned and characterized feline IFNAR2 gene form a cDNA library of feline spleen. This gene was 1,572 bp in length, and coded 523 amino acids (Fig. 1). Compared to IFNAR2 of other species, feIFNAR2 showed the closest similarity to the dog IFNAR2 (GenBank accession no. XM_544861, predicted by automated computational analysis) with 73% similarity at the amino acid level. Its overall structure was also similar to that of other species, not only the complete amino acid length, but also the amino acid length of the extracellular domain and the cytoplasmic domain. The extracellular domain was made of two FBN-III domains, with their characteristic pairs of cysteine residues. Also, as pointed out in the present data, the cytoplasmic region of the feline protein had retained the main signal transduction features of huIFNAR2, including a proline-rich motif and two conserved tyrosine in C-terminus that play important roles in Jak-Stat binding (Domanski and others 1997; Zhao and others 2008).
All type I IFNs exert activity through binding to the same receptor components, IFNAR1 and IFNAR2 (Flores and others 1991; Novick and others 1994; Uzé and others 1995). Several lines of evidence indicated that the feIFNAR-EC showed extensive difference from human IFNAR2-EC (62% amino acid identity). However, a comparison of the sequences of the extracellular region revealed that the basic frame was well-conserved. This portion of the molecule, plus a few other regions of conserved sequence in the two FBN-III domains, might form the primary contact with the counterpart ligands.
Binding of IFN to IFNAR2 is the first step initiating the signal transduction cascade that activates the antiviral state in cells. Here we detected whether both feline IFN-α and IFN-ω interacted with recombinant feIFNAR2 using FACS (Fig. 4B). The results indicated that feIFN-α and feIFN-ω could bind to this protein. Piehler and Schreiber had verified that the non-glycosylated IFNAR2-EC had essentially retained its biological activity and could be used for further biophysical investigations of the type I interferon receptor (Piehler and Schreiber 1999a). Thus the feIFNAR2-EC was expressed in E. coli and tested the binding to IFN (Fig. 6). The ELISA experiment confirmed that the protein could bind to both feIFN-α and feIFN-ω.
Different IFN subtypes have different biological effects. For example, huIFN-α2 and huIFN-β exert comparable specific activities for the antiviral effect against VSV on WISH cells; however, the human IFN-β is much more potent than huIFN-α2 to inhibit the proliferation of these cells (Jaitin and others 2006). In human primary cells, IFN-α8 was shown to be unable to affect T-cell motility, whereas IFN-α2 did (Foster and others 2004). Functional differences among type I IFNs are related to different affinities and kinetics of the interaction with receptor subunits, and antiviral activities are in good agreement with the relative ligand-binding affinities toward IFNAR2 (Piehler and Schreiber 1999b). Although type I IFN binds competitively to the same functional domain, the binding sites for these IFNs on IFNAR2 are different, which could lead to a different orientation of the intracellular domains of IFNAR2 and IFNAR1 in the ternary complex, which could be critical in the pattern of activation of the cellular response to IFN-α2 versus IFN-β. In this study, according to the hotspots on huIFNAR2 for binding to IFNs, three important sites, T75, V76, and M77, were mutated to alanine. The mutational analysis showed that the mutants of T75A and M77A reduced the binding to feIFN-α by 50%. However, these mutants almost did not change the binding to feIFN-ω. The results suggested that feIFN-α and feIFN-ω had distinct binding sites on IFNAR2. Particularly, the residue T75 and M77 were the hotspots for binding to feIFN-α, but not to feIFN-ω. In addition, the mutant of T75A/D82N did not change the binding affinity with feIFN-α, indicating that the mutation of D82 to asparagine could increase the binding affinity. The largest hydrophobic patch on the protein surface is within on IFN-α binding, surrounded by polar and charged residues (Piehler and others 2000). The mutant of M77I also decreased the binding affinity, supposing that the mutant might break the patch between the IFN-α and IFNAR2. Moreover, we found feIFN-ω had broader spectrum of antiviral activity compared with feIFN-α. Although both feIFN-α and feIFN-ω had high antiviral activity in CRFK cells against VSV, feIFN-ω was much more potent than feIFN-α to inhibit the replication of virus in cross-species cells. Thus, to some extent, the present data indicated different binding sites for feIFN-α and feIFN-ω on feIFNAR2, which might result in the different angular orientation of IFN binding to IFNAR2 and lead to the different cellular response to IFNs. However, further studies investigating the differences of feIFN-α and feIFN-ω binding feIFNAR2 are needed.
In conclusion, these experiments provided the primary structures and functions of feline IFNAR2. They indicated that the binding sites of feIFN-α and feIFN-ω on IFNAR2 were different, which maybe related to the antiviral activity of IFN.
Footnotes
Acknowledgments
This work was supported by Ministry of Science and Technology of China (2006DFB32010, 2006DFC30240), National Basic Research Program (973) of China (2005CB523002), and Hundreds of Talents Program of Chinese Academy of Sciences. We thank members of our laboratories who have participated in studies related to this article.