
Abstract
The signal transducers and activators of transcription (STAT)1 and STAT3 genes are specifically activated by phosphorylated STATs 1 and 3, respectively, resulting in large and prolonged increases in the levels of unphosphorylated STATs (U-STATs) in response to interferons (for STAT1) or ligands that activate gp130, such as IL-6 (for STAT3). U-STATs 1 and 3 are transcription factors that drive gene expression by mechanisms distinct from those used by phosphorylated STATs. U-STAT3 drives expression of many proteins not induced by phospho-STAT3, including several that are important in tumorigenesis. U-STAT1 prolongs and increases expression of a subset of proteins induced initially in response to phospho-STAT1, leading to antiviral and immune responses that are long-lived. U-STAT1 levels are also high in some cancers, and the protein products of genes induced by U-STAT1 enhance resistance to DNA damage. Therefore, interferons not only drive short-term expression of proteins that inhibit growth and promote apoptosis and immune surveillance, but also promote long-term expression of proteins that facilitate tumor survival.
Introduction
T
STAT-dependent signaling is tightly controlled. The induced genes include positive and negative regulators that modulate the magnitude and duration of signaling. Several different negative regulators that turn JAK–STAT signaling off soon after activation include protein inhibitor of activated STAT1 and suppressor of cytokine signaling proteins, as well as protein tyrosine phosphatases (Levy and Darnell 2002; Shuai and Liu 2003; Arbouzova and Zeidler 2006). In contrast to the transient activation of STATs that is part of the normal responses to cytokines, persistent STAT activation is frequently associated with malignant transformation. Constitutive activation of STATs, especially STAT3 and STAT5, is found in many human tumors and tumor cell lines, and also in cells transformed in vitro by oncoproteins that activate tyrosine kinase (TK) signaling pathways. Binding of extracellular ligands, including cytokines, growth factors, and hormones (Darnell 1997; Levy and Darnell 2002), to their specific receptors leads to the activation of TKs in addition to JAKs, including receptor TKs, and nonreceptor TKs such as SRC and ABL, which can directly phosphorylate STATs in the absence of ligand-induced receptor signaling (Bowman and others 2000; Bromberg 2001). Constitutively active STAT3 has an important causal role in oncogenesis by promoting uncontrolled growth and survival through uncontrolled expression of genes that include CYCLIND1, C-MYC, BCL-XL, MCL-1, and SURVIVIN.
STATs as Cytokine-Inducible Proteins
Recently, it has been appreciated that STATs 1, 3, and 6 (and possibly others) also play important roles in mediating gene expression without tyrosine phosphorylation (Chatterjee-Kishore and others 2000; Yang and others 2005, 2007; Cui and others 2007; Cheon and Stark 2009). Expression of unphosphorylated STATs (U-STATs) 1 and 3 is greatly increased in response to their activation. The STAT1 gene is strongly activated by phospho-STAT1 (P-STAT1) dimers or ISG factor 3, formed in response to type I or type II IFNs, respectively (Lehtonen and others 1997; Cheon and Stark 2009). STAT2 gene expression is also increased in response to type I or type II IFNs (Lehtonen and others 1997). Similarly, the STAT3 gene is strongly activated by the phosphorylated STAT3 dimers that are formed in response to IL-6 and other ligands that activate the gp130 common receptor subunit. It is remarkable that P-STAT1 increases expression of STAT1 but not STAT3, and vice-versa (Qing and Stark 2004).
Both type I and type II IFNs increase STAT1 expression in many cell types, including normal fibroblasts and mammary epithelial cells, and the newly synthesized STAT1 protein persists for many days after IFN stimulation as U-STAT1, in which Y701 and S727 are not phosphorylated (Cheon and Stark 2009). When the levels of U-STAT1 in normal human fibroblasts are increased, expression of several ISGs is also increased. The same set of ISGs is increased in response to high level of U-STAT1 induced by treatment with IFNs for 48 h in human mammary epithelial cells and fibroblasts. The U-STAT1-induced proteins have immunoregulatory, antiviral, or unknown functions. They include IFI27, IFI44, OAS 1-3, IRF7, MX1, and STAT1 itself (Table 1; Cheon and Stark 2009).
Expression of these genes is driven by increased exogenous expression of STAT1 in normal human BJ fibroblasts. Their expression is also increased by IFN-β or IFN-γ after 6 h, and further increased or stabilized after 48 h, when phospho-STAT1 levels have returned to baseline in treated cells.
IFN, interferon; STAT, signal transducers and activators of transcription.
Mechanisms of STAT Induction by Cytokines
An analysis of regulatory elements in the human STAT1 gene was carried out by Wong and others (2002). Elements required to activate STAT1 expression in response to either type of IFN were found within the gene, rather than in an upstream region. A region of about 1.2 kb, spanning the first exon, the first intron, the second exon and part of the second intron of the human gene was analyzed. The 5′ half of this region contains a 31-bp “IGI” element that is required for function, whereas the 3′ half contains sequences that inhibit function. Even in the absence of IFN treatment, the IGI element binds to IRF1, and increased expression of IRF1 in a human melanoma cell line led to increased STAT1 expression (Wong and others 2002).
Similarly, the STAT3 gene, which contains GAS sequences (Narimatsu and others 2001), is strongly activated by the P-STAT3 that is formed in response to IL-6 and other ligands that activate the gp130 common receptor subunit. The biological role of U-STAT3-driven gene expression in normal physiology was addressed by experiments with genetically altered mice. Narimatsu and others (2001) mutated a GAS element of the endogenous STAT3 promoter. The ability of IL-6 to increase STAT3 expression was abrogated in some tissues but not in others, probably because STAT3-dependent expression of the STAT3 gene can be regulated through additional elements that were not recognized and therefore not mutated. Since complete deletion of STAT3 is embryonic lethal (Takeda and others 1997), it remains to be seen whether mice with complete loss of the STAT3-dependent induction of U-STAT3 expression would have severe defects, as might be expected if the upregulation of U-STAT3 is important for the full physiological functions of the many cytokines that use the common gp130 receptor subunit to phosphorylate STAT3.
U-STAT1 and U-STAT3 Are Transcription Factors
U-STAT1, together with unphosphorylated IRF1, is responsible for constitutive transcription of the LMP2 gene (Chatterjee-Kishore and others 2000). Detailed analysis of the promoter of this gene, together with delineation of how U-STAT1 contributes to its expression, made it clear that U-STAT1 is a transcription factor that is directly responsible for LMP2 expression. The role of U-STATs in gene expression was extended to U-STAT3 by Yang and others (2005). The activation of STAT3 gene expression by P-STAT3 leads to a remarkable increase in the level of U-STAT3, which persists for many days, long after the initial amount of P-STAT3 has disappeared. U-STAT3 drives expression of a set of genes that is mostly distinct from those activated in response to P-STAT3, including oncogenes such as MRAS and MET. Similarly to the situation for U-STAT3, U-STAT1 drives expression of a distinct set of genes, but in this case the genes are also activated by P-STAT1. The U-STAT3 and U-STAT1 responses are completely distinct, with no overlap among the genes that are activated (Yang and others 2005; Cheon and Stark 2009). Further, the STAT1 gene is driven by P-STAT1 and not by P-STAT3; conversely, the STAT3 gene is driven by P-STAT3 and not by P-STAT1 (Qing and Stark 2004). The induction of U-STATs as secondary transcription factors is an element of a more general strategy that cells use to control the duration of expression of specific subsets of the proteins that are induced initially in response to pro-inflammatory cytokines. The newly synthesized proteins, which are induced primarily by P-STATs, carry out initial responses, but some of them cannot be tolerated for very long without causing harm. Therefore, powerful negative feedback mechanisms turn the initial response off after a few hours, and their failure leads to major problems. However, not all of the initially induced genes are turned off rapidly, because it is advantageous to sustain increased expression of a subset of the induced proteins for several days, if these proteins are not deleterious and if they benefit the organism, for example, by sustaining immune responses or antiviral functions in response to IFNs. Thus, a typical response to cytokines involves rapid shutoff of the initial signal, rapid loss of initially induced proteins that can cause harm, but sustained long-term expression of beneficial induced proteins.
How Do U-STAT1 and U-STAT3 Function as Transcription Factors?
U-STATs were initially considered to be latent transcription factors in the cytoplasm, entering the nucleus to induce gene expression only in response to cytokine stimulation. However, STAT1 and STAT3 have been found to be present in nuclei independently of tyrosine phosphorylation, in a cell-type-specific manner (Meyer and others 2002a, 2002b). The nuclear import mechanism of U-STATs is completely distinct from that of phosphorylated STATs. Nuclear import of tyrosine-phosphorylated STAT dimers is dependent on the carrier proteins importins α and β, which interact with nuclear pore proteins. This is an active process requiring energy. On the other hand, U-STATs migrate via direct interaction with nuclear pore proteins independently of metabolic energy. Both pathways operate simultaneously in cytokine-stimulated cells (Meyer and Vinkemeier 2004). U-STAT1 mediates constitutive expression of the LMP2 gene by collaborating with IRF1 (Chatterjee-Kishore and others 2000). U-STAT3 functions as a transcription factor, in part by binding to unphosphorylated nuclear factor κB (NFκB) in competition with inhibitor of NFκB (IκB), driving expression of a small subset of genes that also respond to activated NFκB, such as RANTES, IL-6, and IL-8 (Yang and others 2007). The U-STAT3/U-NFκB complex accumulates in the nucleus with help from the nuclear localization signal of STAT3 (Yang and others 2007). The κB element of the IL-6 gene is driven by canonical NFκB signaling in response to ligands such as TNF or IL-1, leading to the activation of STAT3 in response to secreted IL-6, followed by an increased level of U-STAT3 that sustains initial NFκB-dependent activation of genes such as RANTES for much longer than most of the genes activated by TNF or IL-1. U-STAT6 cooperates with p300 and binds to a consensus STAT6 binding site located within the COX-2 promoter to enhance COX-2 expression (Cui and others 2007). The discovery of these specific mechanisms for how U-STATs mediate gene expression serves as examples for additional mechanisms that have yet to be revealed. We know that a complex of U-STAT3 and U-NFκB drives the induction of some U-STAT3-induced genes, but the remaining induced genes use a different mechanism that remains unknown (Yang and others 2007). By analogy, it seems likely that U-STAT1 might also induce gene expression by using more than one mechanism, using different cofactors and cis-acting elements to activate different target genes. STAT1 and IRF1 bind to each other even in the absence of DNA, and this heterodimer binds to a composite element in the LMP2 promoter that recognizes each monomeric component separately. The ternary complex is stable enough to drive constitutive expression of LMP2 but can be displaced by the more potent P-STAT1 dimer in IFNγ-treated cells. Different complexes of STAT1 with other transcription factors, which may not be very stable in solution, may, nevertheless, form on specific genes, depending for stabilization on binding to specific DNA elements in each promoter and probably also on specific interactions with other bound proteins. Thus, we expect to see several different mechanisms for U-STAT-dependent expression of different genes.
Functions of U-STAT1 in Antiviral and Immune Responses
U-STAT1-induced genes include those whose protein products have immunoregulatory, antiviral, or unknown functions (Table 1; Cheon and Stark 2009). The STAT1 gene is also a target gene of U-STAT1-mediated transcription, and its sequential transcriptional regulation, in response first to P-STAT1 and then to U-STAT1, helps the STAT1 protein to persist for many days in response to IFNs. Similarly, in IFN-treated cells, expression of a subset of ISGs is prolonged selectively by prolonged induction of U-STAT1 as a secondary transcription factor (Fig. 1). Expression of hundreds of ISGs is initiated by P-STAT1, but the transcription of many is not sustained for a long time because the level of P-STAT1 is decreased by the homeostatic actions of negative regulators, such as the phosphatases SHP1 and SHP2 and suppressor of cytokine signaling 1, which are also ISGs (Borden and others 2007). For example, U-STAT1-induced proteins such as OAS1, OAS2, OAS3, and MX1, are essential for complete antiviral responses, and their longer expression helps cells to clear viruses completely. STAT1 has long been known to function as an important component of antiviral and antitumor responses. STAT1-null mice show enhanced susceptibility to bacterial and viral infections, losing responsiveness to both type I and II IFNs (Durbin and others 1996; Meraz and others 1996). The antiviral effects of STAT1 result from expression of ISGs, such as OAS, RNASEL, PKR, MX1, and ISG15 (G1P2) (Borden and others 2007). The corresponding proteins are initially induced by P-STAT1 and remain at high levels in response to U-STAT1 at later times so that cells can clear virus completely.
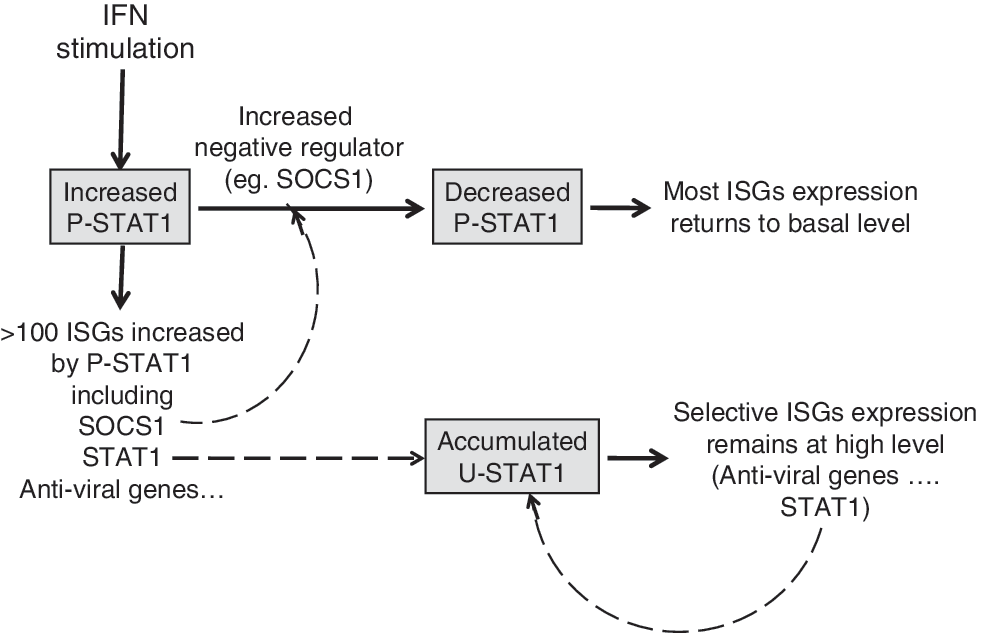
IFN-induced U-STAT1 prolongs expression of selective ISGs. IFN stimulation induces expression of hundreds of ISGs, initially through P-STAT1. The IFN-induced proteins that act as negative regulators, such as SOCS1, inhibit the continued receptor-dependent phosphorylation of STAT1, and expression of most ISGs is thus turned off. The STAT1 gene is also induced in response to IFNs, initially by P-STAT1, and the STAT1 protein accumulates over a period of 8–24 h and remains high for at least several days thereafter. The accumulated U-STAT1 induces expression of a subset of ISGs (Table 2), including STAT1 itself. As a result, the levels of these ISGs remain high for as long as high levels of U-STAT1 protein persist. IFN, interferon; ISG, interferon-stimulated gene; P-STAT1, phospho-STAT1; SOCS1, suppressor of cytokine signaling 1; STAT, signal transducers and activators of transcription; U-STAT, unphosphorylated STAT.
Functions of U-STAT1 in Cancer
STAT1 mediates antitumor functions after its activation by tyrosine phosphorylation. For that reason, it has been thought that high STAT1 levels might result in better antitumor effects by causing more P-STAT1 to be induced in response to IFNs. However, STAT1 is already highly expressed in cancer cells compared to normal cells and in therapy-resistant cancer cells compared to sensitive cells (Perou and others 1999; Khodarev and others 2004; Lesinski and others 2005; Luszczek and others 2010). Functions of some STAT1-induced genes in cancer cells have been investigated, and some have been shown to have pro-metastatic, pro-proliferative, or antiapoptotic properties (Suomela and others 2004; Tahara and others 2005; Pitha-Rowe and Pitha 2007; Hatano and others 2008; Cai and others 2009). STAT1-null or IFN-γ receptor-null mice develop chemically induced and spontaneous tumors more rapidly and frequently, revealing an IFN-mediated tumor surveillance system (Kaplan and others 1998). STAT1 not only regulates immune surveillance, but also increases apoptosis and reduces the proliferation of cancer cells (Bromberg and others 1996, Lee and others 2000). A STAT1-null human cell line (U3A) is not growth-arrested by IFN-α or IFN-γ, in contrast to U3A cells in which STAT1 expression has been restored are (Bromberg and others 1996). Further, lymphocytes derived from STAT1-null mice show decreased apoptosis and increased proliferation in vitro (Lee and others 2000). Certain types of human tumors are unresponsive to IFNs due to defects in the STAT1 activation pathway (Levy and Gilliland 2000). Breast cancer patients with higher levels of phosphorylated and DNA-bound STAT1 show better prognosis and live longer (Widschwendter and others 2002). The mechanism of STAT1-mediated anticancer effects have been extensively investigated, and many pro-apoptotic ISGs (eg, APO2L/TRAIL, FAS, and XIAP) and antiangiogenic ISGs (CXCL9, CXCL10, and CXCL11) have been identified (Borden and others 2007).
However, in contrast to expectations from the results above, the expression level of STAT1 does not influence the response to IFN adjuvant therapy in cancer, and recurrent tumors express higher levels of STAT1 compared to the original tumors (Lesinski and others 2005). Overexpression of STAT1 in recurrent tumors might be caused by IFN treatment, but it has not been understood why increased levels of STAT1 do not confer enhanced anticancer effects. An explanation is suggested by the observation that STAT1 is overexpressed in cancer cells selected for resistance to ionizing radiation and anticancer agents. Khodarev and others (2004) and Luszczek and others (2010) found that several IFN-induced genes are overexpressed in radiation-resistant and chemo-resistant cells. Interestingly, the overexpressed proteins (Table 2) represent only a small subset of the total number of proteins induced by IFNs. These proteins are highly upregulated in resistant cells derived from breast cancers, prostate cancers, gliomas, head and neck squamous carcinomas, and small cell lung carcinomas (Luker and others 2001; Khodarev and others 2004; Tsai and others 2007; Luszczek and others 2010). Many other IFN-induced proteins, for example, APO2L/TRAIL, XAF-1, PRK, and IRF1 (Borden and others 2007) are not upregulated in resistant cells, strongly indicating that IFN signaling is not responsible for STAT1 upregulation in cancer cells.
Increased expression of 24 genes in radio-resistant head and neck cancer cells, compared to the parental cell line (Khodarev and others 2004). N/S R represents the ratios of expression of genes in nu61 cells relative to SCC-61 cells.
Genes found in common in the 2 independent studies.
Genes induced by exogenous overexpression of STAT1 in normal human BJ fibroblasts (Cheon and Stark 2009). Numbers represent the ratios of expression of genes in cells overexpressing wild-type STAT1 (WT) or Y701F STAT1 (YF), relative to cells transfected with empty vector.
Khodarev and others (2007) found that ectopically increased expression of STAT1 can induce a radiation-resistant phenotype. In studying the role of U-STAT1 in IFN-dependent signaling, Cheon and Stark (2009) found that the subset of IFN-induced genes reported to be upregulated in chemo- or radiation-resistant cancer cells are also induced in normal human fibroblasts by exogenous expression of either wild-type STAT1 or Y701F STAT1, which cannot be phosphorylated (Table 2). Therefore, STAT1 is not merely another member of the set of resistance genes but rather is a key transcriptional regulator that induces expression of the other genes. Therefore, the high levels of U-STAT1 seen in many tumors confer a DNA damage-resistant phenotype through the ability of U-STAT1 to drive expression of a relatively small subset of genes. This novel realization has important consequences for the future diagnosis and treatment of cancer. Several major aspects of the role of U-STAT1 in cancer remain to be investigated. What is the mechanism of increased U-STAT1 expression in DNA damage-resistant cancers? What is the mechanism through which U-STAT1 drives gene expression? Which of the induced proteins drive the therapy-resistant phenotype and by what mechanisms, and does increased expression of U-STAT1 contribute to tumorigenesis before treatment?
Weichselbaum and others (2008) extended the scope of the expression signature for resistance to DNA damage to breast cancer and several other tumor types. They also showed that, in addition to STAT1, elevated expression of 2 other genes, ISG15 and IFIT1, could also mediate resistance. The expression levels of 7 genes, including the ones just mentioned, “predicts for efficacy of adjuvant chemotherapy and for local-regional control after radiation.” Khodarev and others (2009) reported that mouse B16F1 tumors grown in syngeneic mice are enriched for cells with STAT1 overexpression and the associated phenotype. Melanoma cell lines with high levels of STAT1 are resistant to doxorubicin, as well as to radiation, when compared to STAT1-low melanoma cell lines, and the STAT1-high cells show impaired caspase 3/7 activation in response to doxorubicin (Khodarev and others 2009). This effect might be caused by increased expression of G1P3 (ISG 6-16), a protein whose expression is elevated by U-STAT1 in normal human BJ fibroblasts and which inhibits TRAIL-induced apoptosis by inhibiting caspase 3 (Cheriyath and others 2007). Luszczek and others (2010) have shown that high STAT1 expression protects small cell lung cancer cells from DNA damage, using comet and phospho-histone H2AX assays. H526 cells, which express relatively low levels of STAT1 and U-STAT1-induced genes, undergo significant DNA damage in response to treatment with a combination of 5-aza-deoxycytidine and MGDC0103, an HDAC inhibitor, but H196 cells, which express high levels of STAT1 and U-STAT1-induced genes, are resistant to DNA damage induced by these agents (Luszczek and others 2010). It has not been reported that any specific U-STAT1-induced protein is directly involved in a mechanism of resistance to DNA damage. Indeed, the roles of the U-STAT1-induced proteins have not been extensively studied in cancer, but some studies have revealed pro-tumorigenic properties. Expression of IFI27 (ISG12) is increased in epithelial cancers, compared to normal skin cells (Suomela and others 2004). Overexpression of the IFITM1 (LEU13) protein has been observed in head-and-neck cancer cell lines, and overexpression of the G1P2 (ISG15) protein has been seen in bladder cancer tissues, correlated with the degree of tumor invasion (Andersen and others 2006; Hatano and others 2008). In each study, IFI27, IFITM1, or G1P2 gene or protein expression was not detected in the corresponding normal tissues or in primary cell controls. Expression of BST2 is also significantly upregulated in bone metastatic breast cancer cell lines and tumor tissues, compared to nonmetastatic cells or tissues (Cai and others 2009). These observations suggest that increased levels of U-STAT1 might participate in oncogenesis as well as resistance to cell death by inducing target genes that increase proliferation, decrease cell death, or increase repair of DNA damage. Why is STAT1 expression elevated in many different cancers? Increased DNA damage in cancer is due to oncogene-induced damage, chromosome instability, and other causes that are intrinsic to tumorigenesis. Therefore, evolving cancer cells must learn to resist the consequences of DNA damage, avoiding normal cellular responses such as cell cycle arrest or apoptosis, or they must repair DNA damage more efficiently, or both. A working hypothesis is that the increase in STAT1 expression in cancers is due to processes intrinsic to tumorigenesis.
Functions of U-STAT3 in Cancer
U-STAT3, which persists for many days after the exposure of cells to IL-6, supports expression of a set of genes that also remain induced for a long time. The high levels of U-STAT3 that accompany the abnormal constitutive activation of STAT3 found in many tumors drives overexpression of several genes that contribute to tumorigenesis, such as SRC (Garcia and others 2001), MET, and MRAS (Yang and others 2005, 2007). Results from cancer cell cDNA arrays indicate that, in tumors of the breast, uterus, and thyroid, MET and MRAS mRNAs are overexpressed in almost all samples analyzed, regardless of the expression levels of STAT3 mRNA (Yang and others 2005). However, in tumors of the colon, stomach, ovary, lung, kidney, and rectum, there is a strong correlation between overexpression of mRNAs encoding MET and MRAS with overexpression of STAT3 mRNA. Thus, MET mRNA is overexpressed in 93% of the tumors in which STAT3 mRNA is overexpressed but in only 29% of the tumors in which it is not, and MRAS mRNA is overexpressed in 74% of the tumors in which STAT3 mRNA is overexpressed but in only 16% of the tumors in which it is not (Yang and others 2005). In cell culture systems, long-term treatment with IL-6 increases total U-STAT3. An array-based analysis of gene expression revealed that the relative levels of more than a thousand mRNAs changed in response to overexpression of STAT3. Some of the genes that respond to U-STAT3 are known to be regulated also by P-STAT3, eg, some oncogenes, C-MYC (Kiuchi and others 1999), C-FOS, C-JUN (Yang and others 2003), and BCL-XL (Niu and others 2002). To analyze the impact of gene expression driven by U-STAT3 in tumors, it will be helpful to determine relative expression of proteins that are driven by the total amount of STAT3 or by P-STAT3, respectively, to help discriminate among different mechanisms in different tumors. For example, if genes that respond to U-STAT3 are expressed in certain cancers but genes that respond to P-STAT3 are not, it would indicate that STAT3 overexpression in these cases is caused by a signal distinct from that generated by P-STAT3. Such information would be useful in seeking appropriate applications for drugs currently in development that target the formation or function of P-STAT3.
New Insights for IFN Therapy
IFNs have been used for the treatment of certain virus infections, multiple sclerosis, and cancer for decades, and their mechanisms of action are still being investigated (Borden and others 2007). The use of IFNs in treating cancer has been limited by significant toxicity and by resistance to therapy. IFNs exert antitumor effects through regulation of hundreds of ISGs, but the functions of many are still unknown. It is very questionable whether all ISGs influence therapies positively when we consider the significant side effects of IFNs. Cheriyath and others (2007) showed that IFN-α2b has a dual role in modulating the balance between myeloma cell survival and death, depending on the duration of treatment, and showed that G1P3 (ISG6-16) antagonizes the effect of TRAIL by inhibiting the intrinsic apoptotic pathway by stabilizing mitochondria. The finding that U-STAT1 and its target ISGs are responsible for the resistance of cancer cells to DNA-damaging therapies provides new insight regarding therapeutic effects of IFNs. Because the IFNs are potent inducers of U-STAT1, their use in therapy is two-edged; P-STAT1 drives antiproliferative and pro-apoptotic effects at early times, but U-STAT1 induces pro-survival effects at late times in response to IFN. If a means could be found to prevent IFNs from increasing the amount of U-STAT1 or to prevent U-STAT1 from inducing gene expression, their therapeutic effects would probably be improved. Therefore, an in-depth understanding of the induction and function of U-STAT1 could lead to more effective therapeutic uses of IFNs.
Footnotes
Acknowledgments
The authors apologize to colleagues whose work was not mentioned due to space limitations. This work was supported by NIH Grant P01 CA62220 to G.R.S.
Author Disclosure Statement
No competing financial interests exist.