
Abstract
Although interferon-γ (IFN-γ) potently inhibits osteoclastogenesis, the suppressive effect is significantly reduced when osteoclast precursors are pre-exposed to the receptor activator of NF-κB (RANK) ligand (RANKL). However, the molecular mechanism underlying the biphasic effects of IFN-γ on osteoclastogenesis remains elusive. Here, we recapitulate the biphasic functions of IFN-γ in osteoclastogenesis in both tissue culture dishes and on bone slices. We further demonstrate that IFN-γ markedly suppresses the RANKL-induced expression of nuclear factor of activated T-cells c1 (NFATc1) in normal, but not RANKL-pretreated bone marrow macrophages (BMMs). Similarly, IFN-γ impairs the activation of the nuclear factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK) pathways in normal, but not RANKL-pretreated, BMMs. These findings indicate that IFN-γ inhibits osteoclastogenesis partially by suppressing the expression of NFATc1 and the activation of the NF-κB and JNK pathways. Moreover, IFN-γ inhibits the RANKL-induced expression of osteoclast genes, but RANKL pretreatment reprograms osteoclast genes into a state in which they can no longer be suppressed by IFN-γ, indicating that IFN-γ inhibits osteoclastogenesis by blocking the expression of osteoclast genes. Finally, the IVVY535–538 motif in the cytoplasmic domain of RANK is responsible for rendering BMMs refractory to the inhibitory effect of IFN-γ. Taken together, these findings provide important mechanistic insights into the biphasic effects of IFN-γ on osteoclastogenesis.
Introduction
I
Osteoclasts are multinucleated giant cells which differentiate from mononuclear cells of the monocyte-macrophage lineage upon stimulation by the macrophage/monocyte-colony stimulating factor (M-CSF) and the receptor activator of NF-κB (RANK) ligand (RANKL) (Boyle and others 2003). RANKL (also known as osteoprotegerin ligand, osteoclast differentiation factor, and TNF-related activation-induced cytokine), a member of the tumor necrosis factor (TNF) superfamily, was identified independently by 2 bone groups (Lacey and others 1998;Yasuda and others 1998) and 2 immunology groups (Anderson and others 1997; Wong and others 1997) in the late 1990s. RANKL plays a pivotal role in bone metabolism by mediating osteoclast differentiation, function, and survival (Lacey and others 1998) and in the immune functions by regulating dendritic cell survival and activation (Wong and others 1997; Josien and others 1999, 2000), T-cell activation (Bachmann and others 1999; Kong and others 1999), B-cell differentiation (Dougall and others 1999; Kong and others 1999), and lymph node development (Dougall and others 1999; Kong and others 1999; Kim and others 2000). Moreover, RANKL is also involved in modulating other physiological processes such as mammary gland development (Fata and others 2000) and thermoregulation in females/fever response in inflammation (Hanada and others 2009).
In bone, RANKL is primarily expressed in osteoblasts and stromal cells (Yasuda and others 1998) and regulates osteoclast differentiation, function, and survival by activating its receptor RANK, a member of the TNF receptor (TNFR) superfamily, in osteoclast precursors and mature osteoclasts. RANK employs 3 TNFR-associated factor (TRAF)-binding sites in its cytoplasmic tail (PFQEP369–373, PVQEET559–564, and PVEQG604–609) (Armstrong and others 2002; Liu and others 2004) to recruit various TRAFs to activate the nuclear factor-κB (NF-κB) and 3 mitogen-activated protein kinase pathways (JNK, c-Jun N-terminal kinase; extracellular signal-regulated kinase; and p38) in osteoclast precursors (Boyle and others 2003; Liu and others 2004; Feng 2005). In particular, PFQEP369–373 has been previously shown to interact with TRAF6 (Ye and others 2002), which plays a crucial role in osteoclast formation and/or function (Lomaga and others 1999; Naito and others 1999). In addition, RANKL induces the expression of nuclear factor of activated T-cells c1 (NFATc1), which has been recognized as a master regulator of osteoclastogenesis (Takayanagi and others 2002; Takayanagi 2007; Aliprantis and others 2008). Importantly, RANK also possesses a TRAF-independent RANK cytoplasmic motif (IVVY535–538) that plays a vital role in osteoclastogenesis by committing bone marrow macrophages (BMMs) to the osteoclast lineage in vitro (Xu and others 2006) and stimulates the osteoclast formation and function in vivo (Kim and others 2009).
Although numerous previous studies demonstrated that IFN-γ is a potent negative regulator of osteoclast differentiation (Takahashi and others 1986; Lacey and others 1995; Fox and Chambers 2000; Kamolmatyakul and others 2001), several groups have shown that IFN-γ exerts a stimulating effect on osteoclastogenesis in vitro and in vivo (Vignery and others 1990; Madyastha and others 2000), raising concerns about the precise role played by IFN-γ in osteoclast biology. Emerging evidence indicates that IFN-γ may play a biphasic role in osteoclastogenesis (Huang and others 2003; Gao and others 2007). Nonetheless, the molecular mechanism by which IFN-γ regulates osteoclastogenesis has not been fully elucidated. In this study, we further investigated the role of IFN-γ in osteoclastogenesis by independently carrying out detailed in vitro studies using mouse BMMs. Our data support the notion that IFN-γ has biphasic functions in osteoclastogenesis. Moreover, we also investigated the molecular mechanism by which IFN-γ exerts the biphasic effects on osteoclastogenesis. We show that IFN-γ exerts the inhibitory effects by suppressing the expression of the NFATc1, impairing the activation of the NF-κB and JNK pathways, and reprogramming of the activation state of osteoclast genes.
Materials and Methods
Chemicals and biological reagents
Chemicals were from Sigma (St. Louis, MO) unless indicated otherwise. Synthetic oligonucleotides were purchased from Sigma-Genosys (Woodlands, TX). Recombinant Mouse IFN-γ was purchased from R&D Systems, Inc. (Minneapolis, MN). Recombinant GST-RANKL was prepared as previously described (McHugh and others 2000). Mouse M-CSF was prepared as culture supernatants from a M-CSF-producing cell line, CMG14-12, kindly provided by Dr. Sunao Takeshita (Takeshita and others 2000). M-CSF concentration was determined by performing MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] assays with M-CSF dependent cell line M-NFS-60 from ATCC (Manassas, VA) using recombinant mouse M-CSF from R&D Systems, Inc. (Minneapolis, MN) as standards. The following antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA): antibodies against nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα) (Cat. No. 9242), phospho-IκBα (2859s), JNK (9252), and phospho-JNK (9251s). Anti-NFATc1 (sc-7294) and anti-TRAF6 (sc-8409) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-human Fas activating antibody (Fas-AB) was purchased from Millipore (Temecula, CA). Anti-human Fas antibody conjugated with phycoerythrin (sc-21730PE) for flow cytometry was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
In vitro osteoclastogenesis assays using primary mouse BMMs
C57BL/6 mice were obtained from Harlan Industries (Indianapolis, IN) and the experiments involving mice were performed in accordance with the regulations of the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee. BMMs were isolated from long bones of young (4–6-week-old) mice and cultured in α-minimal essential medium (α-MEM) containing 10% heat-inactivated fetal bovine serum (FBS) in the presence of M-CSF (220 ng/mL) as previously described (Feng and others 2001). To prepare osteoclasts from BMMs, 5×104 cells per well were cultured in 24-well tissue culture plates in the presence of 44 ng/mL M-CSF and different concentrations of purified GST-RANKL and/or IFN-γ as indicated in individual experiments. The osteoclastogenesis cultures were stained for tartrate resistant acid phosphatase (TRAP) activity using Leukocyte Acid Phosphatase Kit (387-A) from Sigma (St. Louis, MO).
In vitro bone resorption assay
BMMs were plated on bovine cortical bone slices in 24-well plates (5×104/well), then treated with M-CSF (44 ng/mL) with or without RANKL (100 ng/mL) or IFN-γ (1.2 ng/mL) as indicated in individual experiments for 8 days. Bone slices were harvested and cells removed from the bone slices with 0.25
Western blot analysis
BMMs were cultured in α-MEM (serum-free) with M-CSF for 16 h before treatment with RANKL and/or IFN-γ for various times as indicated in individual experiments, then washed twice with ice-cold PBS and lysed in lysis buffer (Cat. No. 9803) from Cell Signaling Technology, Inc. (Beverly, MA), 1×protease inhibitor mixture 1 (P-2850) and 1×protease inhibitor mixture 2 (P-5726) from Sigma were added. Cell lysates was boiled in the presence of sodium dodecyl sulfate (SDS) sample buffer [0.5
Semi-quantitative reverse transcription –polymerase chain reaction
Total RNA was isolated from BMMs using Trizol reagent (Cat. No. 15596-018) from Invitrogen (Carlsbad, CA). 1μg total RNA was reversed transcribed to cDNA with oligo (dT) using the SuperScript® III First-Strand Synthesis System (Cat. No. 18080-051) from Invitrogen (Carlsbad, CA). After completion of reverse transcription, 40 μL H20 was added to the reaction. Using 5 μL for PCR amplification of the matrix metallopeptidase 9 (MMP9), cathepsin K (Ctsk), TRAP, carbonic anhydrase II (Car2), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNAs using the following condition: preheating at 95°C for 2 min, (denaturing 95°C for 30 s, annealing 58°C for 30 s, and extension 72°C for 30 s)×20 cycles for MMP9, Ctsk, TRAP and GAPDH transcripts or×25 cycle for Car2, followed by final extension 72°C for 5 min. PCR was performed with GoTaq Flexi DNA polymerase (Cat. No. M8291) from Promega (Madison, WI) in a 50 μL reaction volume. PCR primers are for GAPDH 5′-ACATCATCCCTGCATCCACTG-3′ and 5′-TCATTGAGAGCAATGCCAGC-3′; for MMP9 5′-CTTCTTCTCTGGACGTCAAATG-3′ and 5′-CATTTTGGAAACTCACACGCC-3′; for TRAP,5′-GCCAAGATGGATTCATGGGTGG-3′ and 5′-CAGAGACATGATGAAGTCAGCG-3′; for Ctsk 5′-GATGCTTACCCATATGTGGGC-3′ and 5′-CATATCCTTTGTTTCCCCAGC-3′; for Car2 5′-AGAGAACTGGCACAAGGACTT-3′ and 5′-CCTCCTTTCAGCACTGCATTGT-3′. Twenty microliters of PCR mixture was loaded on 2% agarose gel for electrophoretic analysis.
Preparation of retrovirus and infection of BMMs
293GPG cells were cultured in DMEM with 10% heat-inactivated FBS supplemented with tetracycline, puromycin, G418, and penicillin/streptomycin. The chimeric receptor constructs, pMX-puro-hFas-RANK [wild type (WT)] and pMX-puro-hFas-PM3 [mutant (Mu)], were generated in previous studies (Xu and others 2004). The retroviral vectors were transiently transfected into 293GPG cells using Lipofectamine Plus Reagent (Invitrogen; Cat. No. 18324-012). After transfection, virus supernatant was collected at 48, 72, and 96 h. BMMs were then infected with virus in the presence of M-CSF (220 ng/mL) and 8 μg/mL polybrene for 24 h. For selection and expansion of transduced cells, cells were further cultured in the presence of M-CSF (220 ng/mL) and 2 μg/mL puromycin for 2 days. Selected cells were subsequently used for various studies. For assays involving the activation of the chimeric receptor, human Fas-AB was added at concentrations as indicated in individual assays.
Flow cytometric analysis
Retrovirally infected BMMs (1×106) were suspended in 200 μL α-MEM containing 10% heat-inactivated FBS in the presence of M-CSF (44 ng/mL). Cells were then blocked with 1 μg of 2.4G2 antibody for 30 min at 4°C. Under dim light, 10 μL human Fas antibody conjugated with phycoerythrin was added to the cell suspension, and cells were incubated for 30 min at 4°C before centrifuging at 2000 rpm for 5 min. Cells were then washed 3 times with 1 mL α-MEM and then suspended in 1 mL α-MEM for cytometric analysis. Flow cytometric analysis was performed using a BD LSR II Flow Cytometer from the Center for AIDS Research at UAB.
Statistical analysis
Bone resorption data are expressed as mean±standard deviation. Statistical significance was determined using Student's t-test and P values <0.05 were considered significant.
Results
IFN-γ exerts biphasic effects on osteoclast formation
The role of IFN-γ in osteoclastogenesis remains controversial (Takahashi and others 1986; Vignery and others 1990; Lacey and others 1995; Fox and Chambers 2000; Madyastha and others 2000; Kamolmatyakul and others 2001). To delineate the precise role of IFN-γ in osteoclastogenesis, we independently and thoroughly carried out numerous assays to examine the effect of IFN-γ on in vitro osteoclastogenesis using primary BMMs (namely, authentic primary osteoclast precursors) from 4- to 6-week-old male C57BL/6 mice. First, BMMs were treated with M-CSF, RANKL and different doses of IFN-γ (0–2.4 ng/mL) for 4 days (Fig. 1A). However, IFN-γ inhibited osteoclastogenesis in a dose-dependent manner and completely blocked osteoclastogenesis at 1.2 ng/mL (Fig. 1A), replicating the conclusion from the previous studies (Takahashi and others 1986; Lacey and others 1995; Fox and Chambers 2000; Kamolmatyakul and others 2001). Next, we determined the effect of IFN-γ on osteoclastogenesis from BMMs that were pretreated by RANKL. BMMs were treated with M-CSF and RANKL for 1 day and then different doses of IFN-γ were added to the cultures to complete the remaining process. As shown in Fig. 1B, a few small osteoclasts formed in the cultures treated with 1.2 ng/mL or 2.4 ng/mL IFN-γ, indicating that the inhibitory effect of IFN-γ on RANKL-mediated osteoclastogenesis is reduced when osteoclast precursors are pretreated by RANKL for 1 day. It was previously shown that pretreatment of BMMs for a longer period can further reduce the inhibitory effect of IFN-γ on RANKL-mediated osteoclastogenesis (Huang and others 2003). To independently validate this finding, we pretreated BMMs with RANKL for 2 days before addition of IFN-γ (Fig. 1C). Our results demonstrate that numerous large osteoclasts formed in the cultures treated with 1.2 ng/mL and 2.4 ng/mL IFN-γ when BMMs were pretreated with RANKL for 2 days (Fig. 1C), recapitulating the previous observation (Huang and others 2003). Conversely, pretreatment of BMMs with IFN-γ for 1 day weakly impairs the ability of BMMs to differentiate into osteoclasts in response to subsequent RANKL stimulation in a dose-dependent manner (Fig. 1D). Moreover, we also repeated the assay shown in Fig. 1A by pretreating BMMs with IFN-γ for 1 day before addition of RANKL (Fig. 1E). The results indicate that pretreatment of BMMs with IFN-γ for 1 day before addition of RANKL more significantly suppresses osteoclastogenesis since IFN-γ at dose of 0.15 ng/mL blocked osteoclastogenesis (Fig. 1E), compared with the inhibitory effect of IFN-γ at this dose in the assay in Fig. 1A. Taken together, these data indicate that IFN-γ can exert biphasic effects on osteoclastogenesis. When IFN-γ is exposed to BMMs before or simultaneously with RANKL, it potently inhibits osteoclastogenesis. In contrast, if IFN-γ is added to BMMs after 1-day RANKL pretreatment, the inhibitory effect of IFN-γ on osteoclastogenesis is impaired.
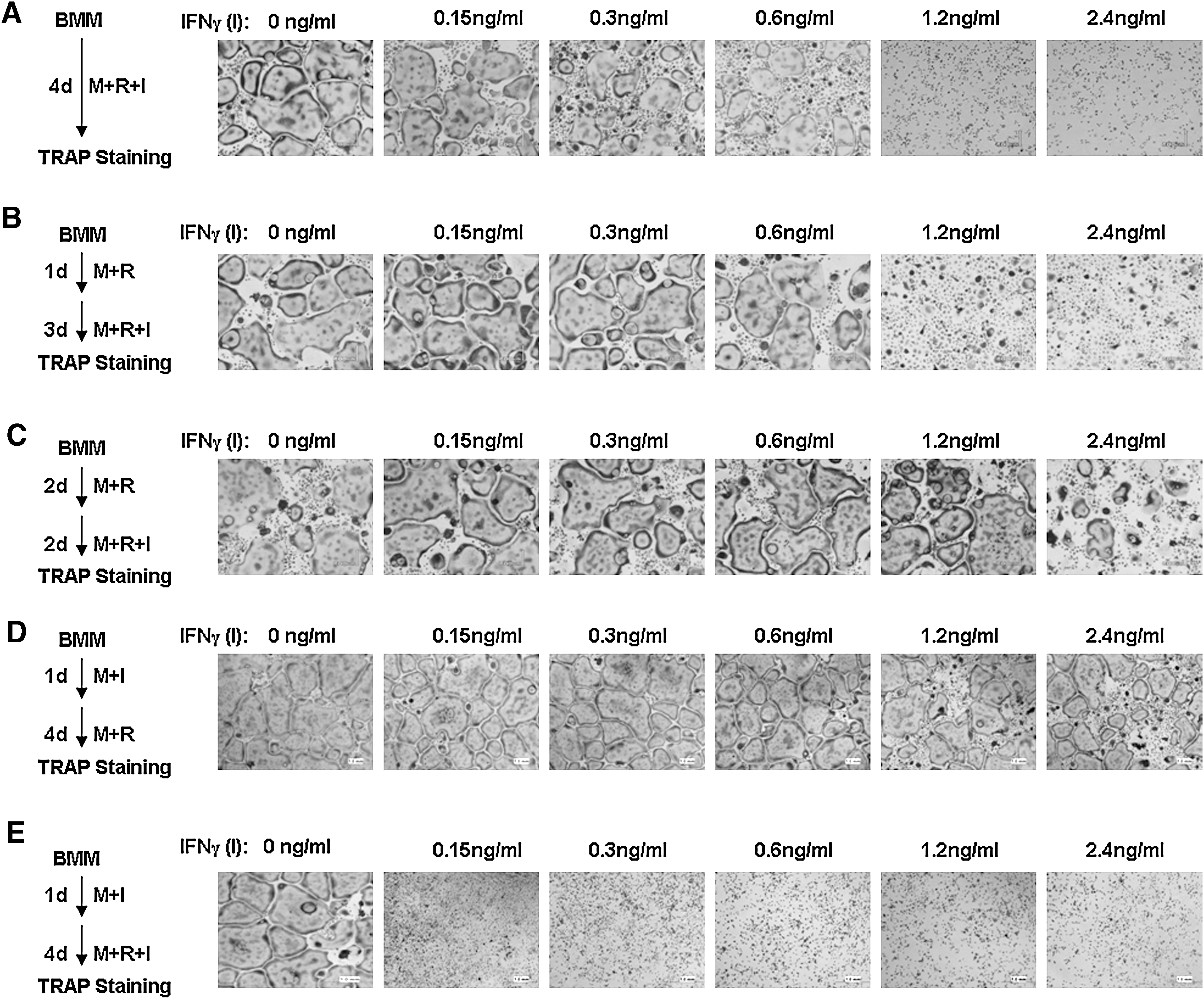
IFN-γ exerts biphasic effects on osteoclastogenesis.
To obtain more physiologically relevant data, we further asked whether we can recapitulate the biphasic effects of IFN-γ on osteoclastogenesis on bone slices. Toward this end, we performed bone resorption assays by plating and culturing BMMs on bone slices (Fig. 2). As a negative control, BMMs grown on bone slices in the presence of M-CSF alone resulted in no bone resorption pits (Fig. 2A). M-CSF and RANKL were sufficient to promote the formation of functional osteoclasts, indicated by the presence of numerous bone resorption pits. In contrast, IFN-γ significantly inhibited the formation of functional osteoclasts, reflected by reduced bone resorption activity (Fig. 2A, B). However, if the cells were first pretreated with M-CSF and RANKL for 2 days and then IFN-γ was added to the culture for the remaining 6 culturing days, the ability of IFN-γ to block the formation of functional osteoclasts on bone slices was significantly impaired (Fig. 2A, B), indicating that pretreatment of BMMs with RANKL can also reduce the inhibitory effect of IFN-γ on osteoclastogenesis on bone slices. This finding further substantiates that IFN-γ plays biphasic roles in osteoclastogenesis.
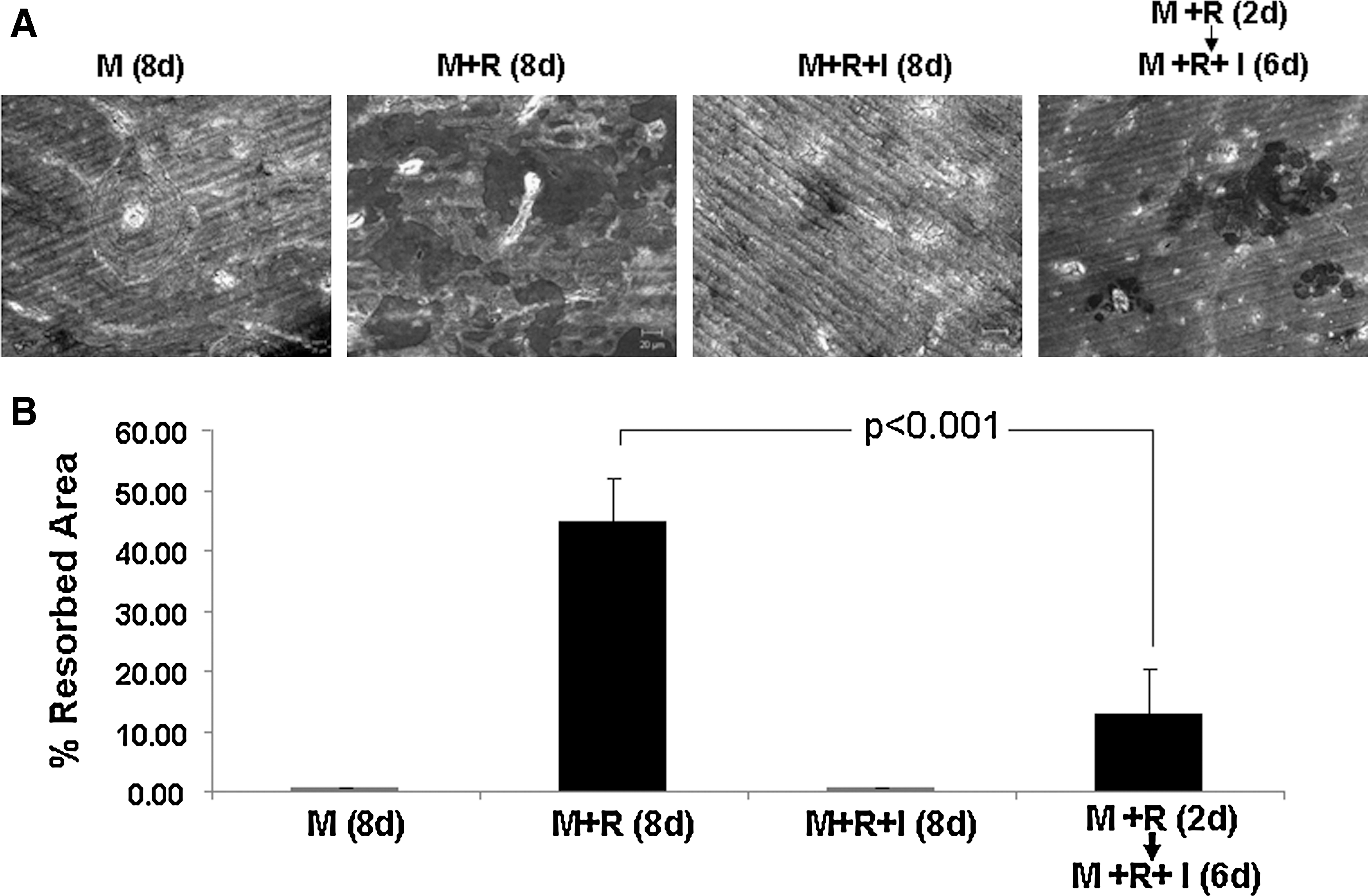
Effects of IFN-γ on osteoclastogenesis on bone slices.
IFN-γ inhibits osteoclastogenesis by impairing the activation of the JNK and NF-κB pathways and suppressing the expression of NFATc1
It was initially shown that IFN-γ inhibits osteoclast formation by inducing degradation of TRAF6 (Takayanagi and others 2000). However, two other groups later failed to replicate this finding (Huang and others 2003; Ji and others 2009). To address this contention, we independently examined the capacity of IFN-γ to promote the degradation of TRAF6. As shown in Fig. 3A, 1-day IFN-γ stimulation exerts no effect on stability of TRAF6 in BMMs. To further address the issue, we repeated the assay with longer IFN-γ treatments and our data demonstrate that the stimulation with IFN-γ as long as 3 days is unable to induce degradation of TRAF6 (Fig. 3B). These findings support the notion that IFN-γ inhibits osteoclast formation by mechanism(s) other than degradation of TRAF6 (Huang and others 2003; Ji and others 2009).
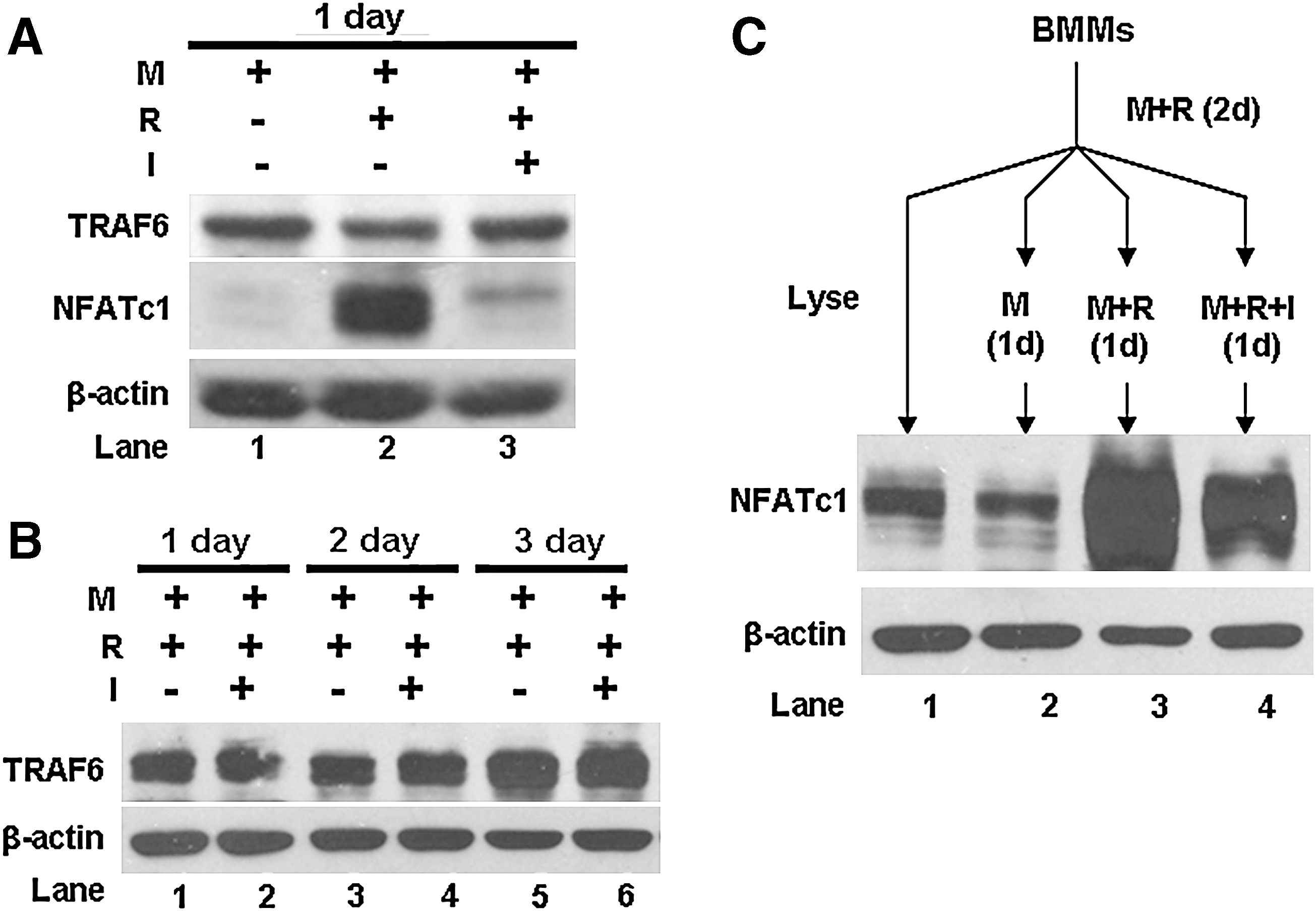
IFN-γ exerts no effect on TRAF6 stability but regulates differently the expression of NFATc1 in normal BMMs and RANKL-pretreated BMMs.
To elucidate the precise mechanism by which IFN-γ suppresses osteoclastogenesis, we examined the effect of IFN-γ on the expression of NFATc1, which is a master regulator of osteoclastogenesis (Takayanagi and others 2002; Takayanagi 2007; Aliprantis and others 2008). To this end, we treated BMMs with RANKL for 24 h in the absence or presence of IFN-γ and the expression of NFATc1 was assessed by western blot analysis. As shown in Fig. 3A, while RANKL dramatically up-regulated NFATc1 expression (lane 2), the addition of IFN-γ almost completely abrogated the RANKL-induced NFATc1 expression (lane 3). Given that we and others have demonstrated that 2-day RANKL pretreatment significantly reduces the inhibitory effect of IFN-γ on osteoclastogenesis (Fig. 1C) (Huang and others 2003), we further examined whether RANKL pretreatment abrogates the IFN-γ-mediated inhibitory effect on the expression of NFATc1. The data demonstrate that while NFATc1 expression induced by 2-day RANKL pretreatment does not significantly decrease during the following 1-day M-CSF-only stimulation period (lanes 1 and 2, Fig. 3C), RANKL treatment further enhances NFATc1 expression after 2-day RANKL pretreatment (lanes 2 and 3, Fig. 3C). Moreover, IFN-γ slightly inhibits RANKL-induced additional NFATc1 expression after 2-day RANKL pretreatment (lanes 3 and 4, Fig. 3C). Taken together, these findings indicate that IFN-γ inhibits osteoclastogenesis in part by suppressing the RANKL-induced activation of NFATc1.
Previously it was shown that IFN-γ inhibits osteoclastogenesis by suppressing the RANKL-induced activation of the NF-κB and JNK signaling pathways in osteoclast precursors (Takayanagi and others 2000). To further delineate the molecular mechanism by which IFN-γ suppresses osteoclastogenesis, we also examined the effect of IFN-γ on RANKL-induced activation of the NF-κB and JNK pathways in BMMs. As shown in Fig. 4, BMMs were treated with RANKL for 5 or 10 min without or with IFN-γ and the activation of the NF-κB and JNK pathways was assessed. Our results show that IFN-γ impaired the activation of the NF-κB and JNK signaling pathways (compare lanes 2 vs. 4, and lanes 3 vs. 5, Fig. 4). Moreover, we also examined whether RANKL pretreatment abrogates the IFN-γ-mediated inhibitory effect on the activation of the NF-κB and JNK signaling pathways. Our data demonstrate that that IFN-γ can no longer exert the inhibitory effect on the activation of the NF-κB and JNK signaling pathways once BMMs are pretreated with RANKL for 2 days (compare lanes 7 vs. 9, and lanes 8 vs. 10, Fig. 4). These findings indicate that IFN-γ inhibits osteoclastogenesis also by interfering with the activation of the JNK and NF-κB pathways.

IFN-γ inhibits RANKL-mediated activation of the NF-κB and JNK pathways in normal BMMs but not in RANKL-pretreated BMMs. Normal BMMs or BMMs pretreated with RANKL for 2 days were cultured with serum-free media for 16 h before treatment with RANKL for 0, 5, or 10 min in the absence or presence of IFN-γ. Activation of the NF-κB and JNK pathways was assessed as phosphorylation of IκB and JNK using western analysis with antibodies against phospho-IκB (p-IκB) and phospho-JNK (p-JNK). The same volume of lysates was run and then probed with antibodies against IκB and JNK as loading control. The assays were independently repeated 3 times. A representative set of data is shown. Band densities were determined as described in the Materials and Methods section and presented as p-IκB/IκB (total) and p-JNK/JNK (total) ratios below the images. NF-κB, nuclear factor-κB; JNK, c-Jun N-terminal kinase.
IFN-γ inhibits osteoclastogenesis by preventing the activation of the expression of osteoclast genes
RANKL promotes osteoclast differentiation by altering the expression of a large number of genes (Cappellen and others 2002; Ishida and others 2002; Rho and others 2002). Among them are those encoding MMP9, Car2, Ctsk, and TRAP. To further decipher the molecular mechanism by which IFN-γ inhibits osteoclastogenesis, we examined the effect of IFN-γ on the expression of the four osteoclast genes (Fig. 5). BMMs were treated with RANKL for 1, 2, or 4 days in the absence or presence of IFN-γ. Although RANKL significantly increased the expression of these genes with time, the presence of IFN-γ in the cultures completely blocked the activation of the expression of these genes (Fig. 5A), indicating that IFN-γ inhibits osteoclastogenesis by preventing the activation of the expression of osteoclast genes.
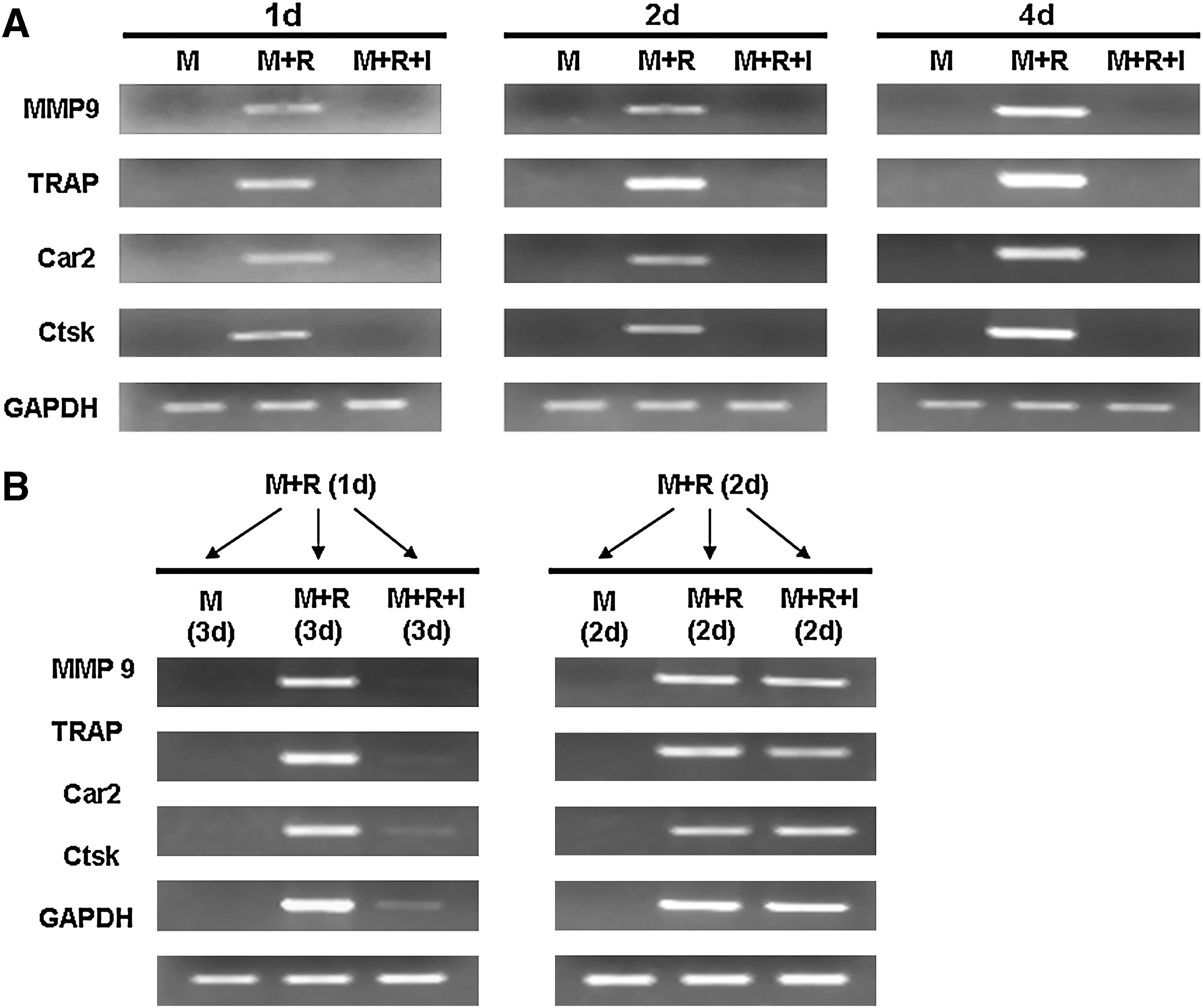
IFN-γ inhibits RANKL-mediated expression of osteoclast genes in normal BMMs but not in RANKL-pretreated BMMs.
We then examined whether RANKL pretreatment can eliminate the inhibitory effect of IFN-γ on the expression of the 4 osteoclast genes (Fig. 5B). BMMs were pretreated with RANKL for 1 or 2 days and then the cultures were continued with M-CSF alone, M-CSF and RANKL, or M-CSF and RANKL plus IFN-γ. Although IFN-γ still inhibited the expression of the MMP9, TRAP, Car2, and Ctsk genes in BMMs pretreated with RANKL for 1 day, it failed to inhibit the expression of these genes in BMMs pretreated with RANKL for 2 days. The findings indicate that while IFN-γ inhibits osteoclastogenesis by preventing the activation of the expression of osteoclast genes, the RANKL pretreatment renders BMMs refractory to the inhibitory effect of IFN-γ by altering osteoclast genes into a state in which IFN-γ is no longer able to suppress the activation of the expression of osteoclast genes.
The RANK IVVY535–538 motif plays a critical role in rendering osteoclast precursors refractory to the inhibition of IFN-γ
A TRAF-independent RANK cytoplasmic motif (IVVY535–538) has been shown to play a crucial role in osteoclastogenesis by priming BMMs to the osteoclast lineage in vitro (Xu and others 2006). Thus, we investigated whether this IVVY motif also plays a critical role in rendering osteoclast precursors refractory to the inhibition of IFN-γ. To do so, we took advantage of 2 chimeric receptors that were previously constructed and validated (Xu and others 2004, 2006). Briefly, the chimeras were prepared by linking the external domain of human Fas to the transmembrane and cytoplasmic domains of normal mouse RANK (WT) or RANK bearing inactivating mutations in the IVVY motif (Mu; Fig. 6A) (Xu and others 2004). These chimeras can be activated by a human-specific Fas-AB to exclusively activate RANK without affecting endogenous mouse Fas (Xu and others 2004). Flow cytometric analysis showed that WT and Mu were expressed at comparable levels on the surface of infected cells (Fig. 6B). WT-expressing or Mu-expressing cells were treated with M-CSF and Fas-AB for 2 days before the cultures were switched to M-CSF alone, M-CSF and RANKL, or M-CSF and RANKL plus IFN-γ for 4 days (Fig. 6C). WT-expressing cells formed a few small osteoclasts in the presence of M-CSF alone after 2-day Fas-AB treatment. This is likely to result from the overexpression of the WT chimeric receptor in a few cells in the infected BMM population and it is well known that the overexpression of TNF family members can lead to self-activation (Inoue and others 2000). As expected, Fas-pretreated BMMs formed large multinucleated osteoclasts in response to subsequent M-CSF and RANKL stimulation for 4 days. Moreover, addition of IFN-γ to the culture still resulted in significant osteoclast formation from WT BMMs. Thus, this finding has recapitulated our previous observations that the chimera containing the external domain of human Fas to the transmembrane and cytoplasmic domains of normal mouse RANK (WT) can be activated by Fas-AB to transmit RANK signaling pathways (Xu and others 2004). Mu-expressing cells formed no osteoclasts in the presence of M-CSF alone after 2-day Fas-AB treatment, but the cells formed large multinucleated osteoclasts in response to subsequent M-CSF and RANKL stimulation for 4 days (Fig. 6C). Importantly, IFN-γ was able to inhibit formation of osteoclasts from Mu-expressing BMMs after 2-day Fas-AB treatment. These results indicate that the RANK IVVY535–538 motif plays a critical role in rendering osteoclast precursors refractory to the inhibition of IFN-γ.
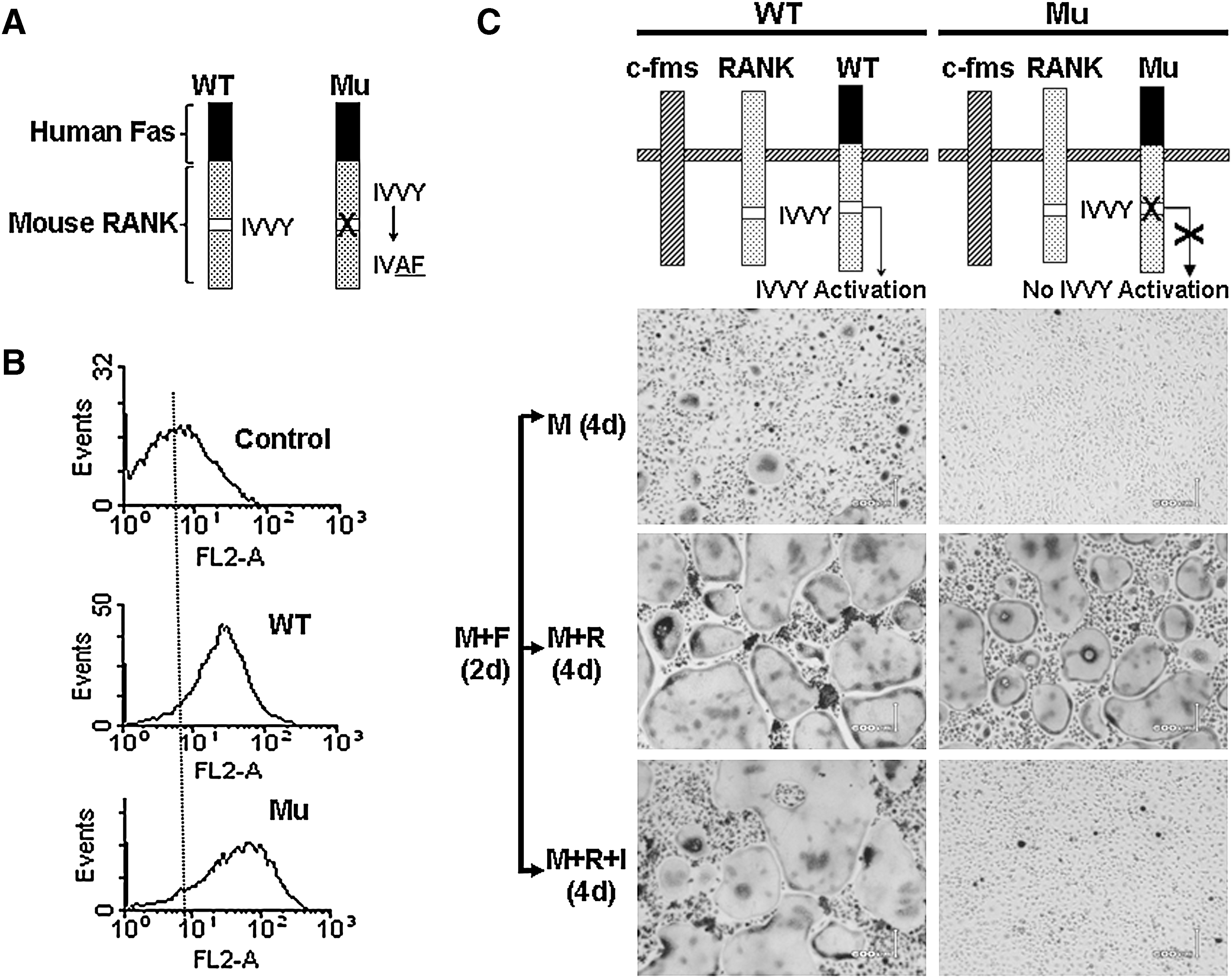
The RANK IVVY535-538 motif plays a critical role in rendering BMMs refractory to the inhibition of IFN-γ.
Discussion
An inhibitory effect of IFN-γ on bone resorption was initially discovered almost a quarter of century ago in an in vitro neonatal mouse bone resorption assay (Gowen and Mundy 1986). Numerous subsequent studies further demonstrated that IFN-γ exerts the suppressive effect on bone resorption primarily by blocking osteoclastogenesis (Takahashi and others 1986; Lacey and others 1995; Fox and Chambers 2000; Kamolmatyakul and others 2001). However, the physiological significance of the inhibitory role of this Th1 cytokine in osteoclastogenesis had remained elusive until the unraveling the RANKL/RANK system. In the late 1990s, RANKL was almost simultaneously discovered by bone biologists (Lacey and others 1998; Yasuda and others 1998) and immunologists independently (Anderson and others 1997; Wong and others 1997). Although bone researchers demonstrated that RANKL is a potent stimulator of bone resorption via promoting osteoclast formation, function, and survival (Lacey and others 1998; Yasuda and others 1998), immunologists showed that RANKL is also an important regulator of immune functions such as dendritic cell survival and activation (Wong and others 1997; Josien and others 1999, 2000), T-cell activation (Bachmann and others 1999; Kong and others 1999), and B-cell differentiation (Dougall and others 1999; Kong and others 1999). In particular, it was shown that RANKL expression is up-regulated in activated T cells to promote dendritic cell survival and activation (Wong and others 1997; Josien and others 1999, 2000). This raised a question: how is extensive bone resorption prevented in the constant immune response involving the activation of T cells? Not long after the unraveling of the RANKL/RANK system, it was revealed that IFN-γ, produced by activated T cells, plays an important role in preventing abnormal bone resorption during normal T-cell–mediated immune response by counteracting the stimulatory effect of T-cell–produced RANKL on osteoclastogenesis (Takayanagi and others 2000).
Activated T cells are present in a number of chronic inflammatory conditions such rheumatoid arthritis (Cope and others 2007) and periodontitis (Taubman and Kawai 2001), which are characterized by extensive bone loss. So, this leads to another intriguing issue: why is IFN-γ, produced by activated T cells, unable to prevent excessive bone resorption by blocking the RANKL-induced osteoclastogenesis in these pathological conditions? Intriguingly, a study showed that IFN-γ failed to inhibit osteoclastogenesis from a macrophage cell line or spleen cells which were pretreated by RANKL for 2 days (Huang and others 2003). Our first objective of the present study was to further validate the finding by independently examining the regulatory role of IFN-γ in osteoclastogenesis using BMMs, which represent authentic primary osteoclast precursors. Moreover, we also demonstrated that osteoclasts derived from RANKL-pretreated BMMs are functional in the presence of IFN-γ. These findings may provide an answer to the question of why IFN-γ is unable to prevent excessive bone resorption in rheumatoid arthritis and periodontitis. In healthy individuals, RANKL levels in the bone marrow or RANKL expression levels on stromal cell/osteoblasts are low and thus BMMs are pre-exposed to low levels of RANKL. Thus, when these osteoclast precursors are recruited into inflammatory sites rich in activated T cells, IFN-γ is able to prevent formation of osteoclastogenesis. However, in certain individuals, RANKL levels in the bone marrow may be elevated and BMMs are pretreated by RANKL in the bone marrow before they are recruited to inflammatory sites. These pretreated osteoclast precursors are able to form osteoclasts in presence of IFN-γ. Consistent with this notion, RANKL expression levels are elevated in bone marrow cells in postmenopausal women (Eghbali-Fatourechi and others 2003) and periodontal bone loss is associated with postmenopausal osteoporosis. Moreover, it has been also shown that stromal cells/osteoblasts from aged mice (C57BL/6) express higher levels of RANKL (Cao and others 2003) and age is one of key risk factors for periodontal bone loss.
Interestingly, it is noted that although osteoclasts from 2-day RANKL pretreatment followed by addition of IFN-γ for the remaining 6 days exhibited much higher bone resorption activity than those from 8-day RANKL treatment control (Fig. 2), osteoclast formation in tissue culture dishes in response to 2-day RANKL pretreatment followed by addition of IFN-γ for the remaining 2 days is similar to that stimulated by 4 day RANKL treatment (Fig. 1C). This discrepancy may reflect the fact that the formation of osteoclasts in tissue culture dishes is primarily signified by only few features of osteoclasts such as TRAP expression and cell fusion (leading to a change in cell size and multinucleation), osteoclast differentiation involves the regulation of a large number of genes and many other cellular changes. Our data suggest that while IFN-γ does not significantly affect cell fusion, it may considerably impair differentiation of BMMs into functional osteoclasts by inhibiting the expression of osteoclast genes required for bone resorption as shown in Fig. 5.
Our second goal was to investigate the molecular mechanism by which IFN-γ exerts the biphasic effects on osteoclastogenesis. It was originally shown that IFN-γ inhibits osteoclast formation by causing degradation of TRAF6, which further leads to impaired activation of the JNK and NF-κB pathways (Takayanagi and others 2000). This finding was subsequently challenged by 2 studies demonstrating that IFN-γ exerts no effect on the stability of TRAF6 (Huang and others 2003; Ji and others 2009). We also failed to observe an effect of IFN-γ on the stability of TRAF6 (Fig. 3). However, our data replicated the previous finding that IFN-γ suppresses the activation of the JNK and NF-κB pathways (Takayanagi and others 2000). More importantly, we have further demonstrated that IFN-γ-mediated inhibitory effect on the RANKL-induced activation of the JNK and NF-κB pathways is abrogated in BMMs pretreated by RANKL for 2 days, indicating that IFN-γ exerts the biphasic effects on osteoclastogenesis in part by suppressing the activation of the JNK and NF-κB pathways.
NFATc1 has been established as a master regulator of osteoclastogenesis (Takayanagi and others 2002; Takayanagi 2007; Aliprantis and others 2008). Recently, we have demonstrated that lipopolysaccharide plays a bifunctional role in osteoclastogenesis by regulating the expression of NFATc1 (Liu and others 2009). In our present study, we demonstrate that IFN-γ potently inhibits RANKL-induced NFATc1 expression in BMMs (Fig. 3A), indicating that IFN-γ inhibits osteoclastogenesis in part by suppressing the RANKL-induced activation of NFATc1 expression. Moreover, we show that NFATc1 expression levels in BMMs pretreated with RANKL for 2 days do not significantly decline with time (Fig. 3C), revealing that NFTAc1 is relatively stable in BMMs. This finding explains why IFN-γ exhibits reduced ability to inhibit osteoclastogenesis from RANKL-pretreated BMMs, namely, NFATc1 levels in RANKL-pretreated BMMs are largely sufficient to support osteoclastogenesis. Interestingly, we also demonstrate that continued RANKL treatment after 2-day RANKL pretreatment further enhances NFATc1 expression and IFN-γ only moderately inhibits the RANKL-induced additional NFATc1 expression (lanes 3 and 4, Fig. 3C). These data support the notion that once the NFATc1 gene is primed by RANKL, it becomes somewhat refractory to the inhibitory effect of IFN-γ.
Moreover, microarray studies have revealed that RANKL mediates osteoclastogenesis ultimately by regulating the expression of a large number of genes (Cappellen and others 2002; Ishida and others 2002; Rho and others 2002), including those encoding well-known osteoclast markers such as MMP9, Car2, Ctsk, and TRAP. Our data demonstrate that although IFN-γ potently inhibits the expression of the four osteoclast genes, it is no longer able to suppress the expression of these osteoclast genes in osteoclast precursors pretreated by RANKL for 2 days. Based on these findings, we conclude that IFN-γ inhibits osteoclastogenesis by preventing the activation of the expression of osteoclast genes.
Finally, we determined whether an TRAF-independent RANK cytoplasmic motif (IVVY535–538) is specifically involved in rendering osteoclast precursors refractory to the inhibition of IFN-γ since we previously showed that this motif plays an essential role in osteoclastogenesis by priming BMMs to the osteoclast lineage in vitro (Xu and others 2006). To this end, we used a chimeric receptor approach (Fig. 6). Our data indicate that the activation of the wild type chimeric receptor (WT) for 2 days efficiently rendered BMMs refractory to the inhibitory effect of IFN-γ as these cells were able to form osteoclasts in the presence of M-CSF, RANKL, and IFN-γ (Fig. 6C). In contrast, 2-day activation of the chimeric receptor bearing an inactivating mutation on the IVVY motif (Mu) failed to make BMMs refractory to the inhibitory effect of IFN-γ since the cells did not differentiate into osteoclasts in response to M-CSF, RANKL, and IFN-γ treatment (Fig. 6C). Although the precise mechanism by which the IVVY motif mediates the refractory effect remains to be determined, a recent study has demonstrated that that the IVVY motif regulates osteoclastogenesis by engaging Vav3 indirectly via an unknown adaptor protein (Kim and others 2009). Thus, one potential mechanism may involve the reorganization of cytoskeleton by the IVVY motif-mediated signaling, resulting in the constitutive activation of the RANK signaling important for osteoclastogenesis. Future studies aimed at delineating the molecular mechanism by which the IVVY motif mediates the refractory effect of IFN-γ on osteoclastogenesis may provide crucial mechanistic insights into the molecular mechanisms of osteoclastogenesis.
In summary, our key findings are that (a) IFN-γ exerts inhibitory effects on osteoclastogenesis by suppressing the activation of the JNK and NF-κB pathways and suppressing the expression of NFATc1, (b) the inhibition of osteoclastogenesis by IFN-γ is ultimately regulated at the gene expression levels, and (c) the RANK IVVY535–538 motif plays a critical role in rendering osteoclast precursors refractory to the inhibition of IFN-γ. Our current findings provide important new insights into the molecular mechanism by which IFN-γ regulates osteoclastogenesis. Nonetheless, these observations have also raised additional questions to be addressed in the future. First, it was previously believed that IFN-γ suppresses the activation of the JNK and NF-κB pathways through IFN-γ-induced degradation of TRAF6 (Takayanagi and others 2000). Since we and others have demonstrated that IFN-γ has no effect on the stability of TRAF6, it is likely that IFN-γ impairs the activation of the JNK and NF-κB pathways by a mechanism(s) independent of TRAF6. Second, while IFN-γ potently inhibits the RANKL-mediated NFATc1 expression, the IFN-γ-imposed inhibition on the activation of NFATc1 expression by RANKL was markedly reduced in BMMs pretreated by RANKL. The precise mechanism underlying the effects of IFN-γ on NTAFc1 expression remains to be elucidated. Hence, future studies aimed at addressing these issues will not only provide additional insights into the molecular mechanism by which IFN-γ regulates osteoclastogenesis but may also reveal novel therapeutic targets for treating and preventing bone loss in various pathological conditions.
Footnotes
Acknowledgments
This work was supported by a grant (No. AR47830 to X.F.) from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, a Within Our Reach Innovative Basic Research grant from Research and Education Foundation of American College of Rheumatology (to X.F.), a pilot grant from the Center for Metabolic Bone Disease at UAB (to S.W.), and the National Scholarship for Building High Level University from Chinese Ministry of Education (to J.C.).
Author Disclosure Statement
No competing financial interests exist.